转录组测序1-测序原始数据说明
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了转录组测序1-测序原始数据说明相关的知识,希望对你有一定的参考价值。
参考技术A 转录组测序是最常用的组学实验,对全谱基因定量,找到差异表达基因。RNAseq涉及到原始数据,数据质控,基因组比对,差异基因鉴定,差异基因功能富集分析,重要基因如转录因子激酶的靶基因预测等,我们用10讲的时间,全面讲解转录组测序报告,及在上百个项目中遇到的近百个常见问题。本节视频是第1讲,从测序原始下机数据开始讲起。样品制备到产出测序数据过程,是RNA提取建库和测序的试验环节。在使用Illumina系列的仪器进行测序后,测序下机数据以fastq文件存储,称为raw data(或raw reads),记录了转录组RNA序列的碱基组成信息。
(1) 如何查看fastq文件;
(2) fastq文件的基本格式,每一行的主要信息;
(3) 如何在fastq文件中评估碱基质量,代表碱基质量的ASCII值和单碱基测序错误率之间怎样换算;
(4) N碱基代表什么,常说的Q20、Q30等与测序质量间存在怎样的关系,一般它们多少时代表测序质量是合格的;
(5) 为什么一个样本对应R1和R2两个fastq文件,什么是双端测序。
视频教程:
bilibili超清视频链接: https://www.bilibili.com/video/BV1TJ411V7av
干货分享转录组测序数据挖掘思路&分析方法大放送
近几年,随着高通量测序的普遍性及适用性不断提高,高通量测序数据飞速增长,公共数据已经达到了PB级别,并伴随着项目同质化的问题,所以项目的个性化分析和数据挖掘成为了科学研究和文章发表所必需的。
我们拿到测序数据结果之后,如何针对自己的方案设计与研究思路对数据进行深入挖掘,得到其中的关键信息与结果呢?今天小编就跟大家分享一下转录组测序数据如何进行深入挖掘。
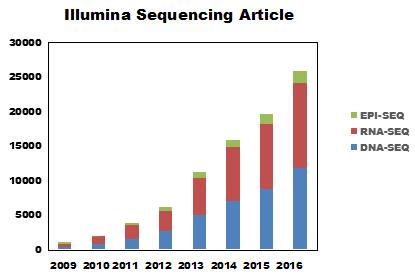

数据挖掘思路
经典思路1:基于差异表达基因
众所周知,差异表达基因(DEG)筛选是转录组测序分析的核心基础,而标准分析结果只包含了有限的差异分组和固定的筛选参数,不能完全满足数据深度挖掘的需求,尤其是样本数较多、试验设计较复杂、DEG过多或过少的时候,因此需要在标准分析的基因表达量基础上,设置差异比较分组、合适的分析软件或筛选参数阈值,获取符合方案设计的DEG结果。
在获得组间或者个体间DEG之后,通过进一步的挖掘锁定核心DEG:
DEG功能分析
对DEG进行功能注释与富集分析,筛选注释、富集到与关注表型或性状等相关的功能或途径的DEG。比如研究肿瘤发生、发展机制,肿瘤样本与健康对照之间的DEG中,注释到细胞增殖、恶性疾病的细胞分化、发育过程等相关的功能或途径(GO term、KEGG pathway等)的基因,可能是肿瘤发生与发展中起重要作用的关键候选基因。
组间DEG维恩图分析
根据组间共有DEG或组内特有DEG缩小候选关键基因范围。比如研究肿瘤发展过程中的分子机制,相比于健康对照组,在不同发展时期的肿瘤样本中均差异表达的基因,可能在整个发展过程中都发挥重要作用。
DEG蛋白互作分析
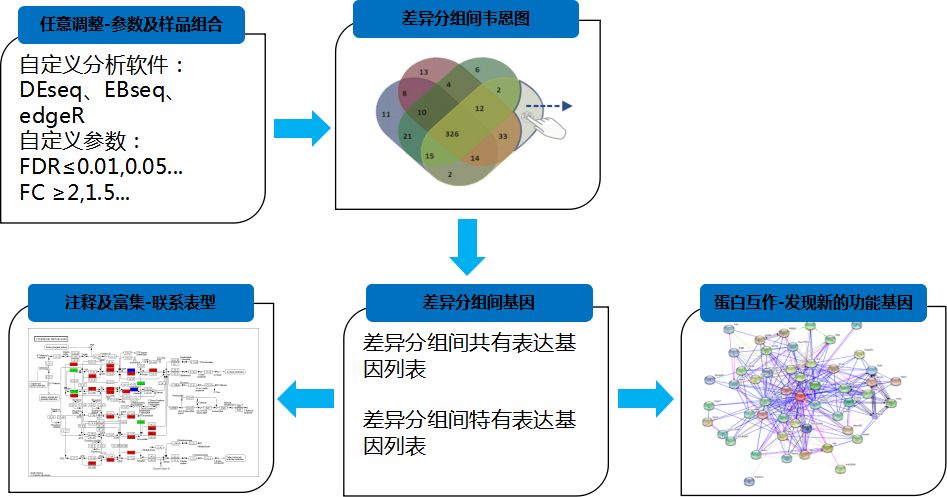
经典思路2:基于基因表达模式
参与同一生物过程的基因通常受同一个调控系统的控制,即参与同一生物过程中的基因有着相似或相同的变化规律。因此,通过基因在不同样品中的表达模式的分析,获得表达模式相近的基因模块,结合功能注释与富集分析,或者与性状关联分析,进而联系表型筛选关键基因模块。一般有以下两种类型:
基因共表达趋势分析
写出好标题
用于识别差异表达基因的共表达模式(common expression patterns),适用于2个以上的样本,尤其是时序样本。
分析各样品间mRNA表达丰度的不同变化模式,将相同表达趋势的mRNA划分为一个数据集,并对该数据集作表达模式图。所用距离测度为欧氏距离,聚类方法为K均值聚类或层次聚类,K均值聚类最终会数据划分为K个类。
WGCNA
1)WGCNA
WGCNA(Weighted gene co-expression network analysis )是构建基因共表达网网络的主要方法。适用于5组以上(每组至少3个重复)样品的数据。
WGCNA算法首先假定基因网络服从无尺度分布,并定义基因共表达相关矩阵、基因网络形成的邻接函数,然后计算不同节点的相关系数,并据此构建分层聚类树,该聚类树的不同分支代表不同的基因模块,模块内基因共表达程度高,而分属不同模块的基因共表达程度低。最后,探索模块与特定表型或疾病的关联关系,最终达到鉴定基因网络的目的。
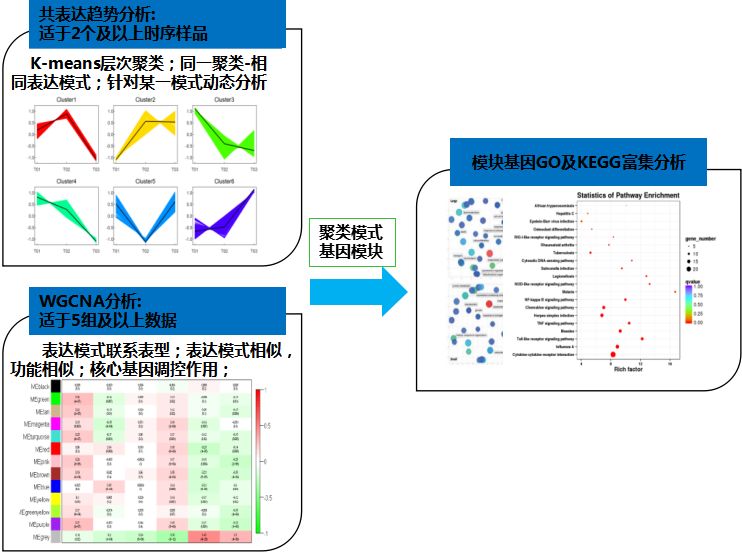
数据挖掘方法
当我们整理好挖掘思路、设计好挖掘方案,就要开始着手分析了,一般来说不外乎两种方法:
A:自己分析。对于有生信背景,熟悉这几种软件或算法,会写代码的童鞋,想来并不算难事,然而对于大部分人来说,这种方法难度系数还是比较高;
B:公司分析。这一方法相比于自己分析而言,把自己的思路与方案均抛之公司,省心又省力。然而沟通效率和思路再现中存在的问题是客观存在的,生信工程师做到完全理解、实施分析,并根据结果与预期做适当调整,以及体现其中生物学意义,又谈何容易。此外,时间上也比不上自己分析来得快。
这两种方法各有优势,但不足之处也不言而喻,今天要跟大家介绍的是第三种方法,一种无需生信知识、省时、高效的方法--百迈客云平台可视化的数据分析APP。
我们之前也介绍过()数据分析APP不仅可以实现快速标准分析,还可以进行个性化分析,可自定义参数方案,并支持界面式数据挖掘,分析得到的图、列表等结果都可以下载到本地电脑,亦可通过保存至研究进展储存在云上。
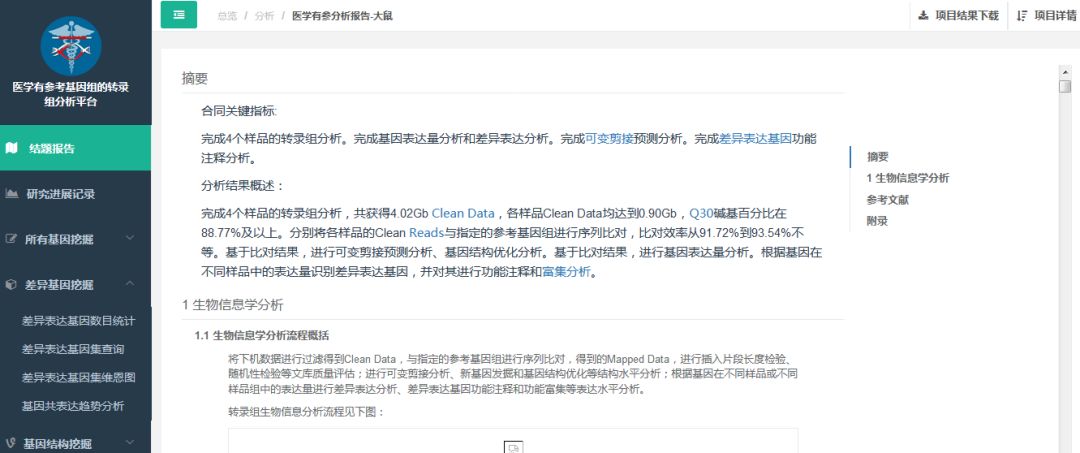
转录组分析APP
个性化分析有三大模块:
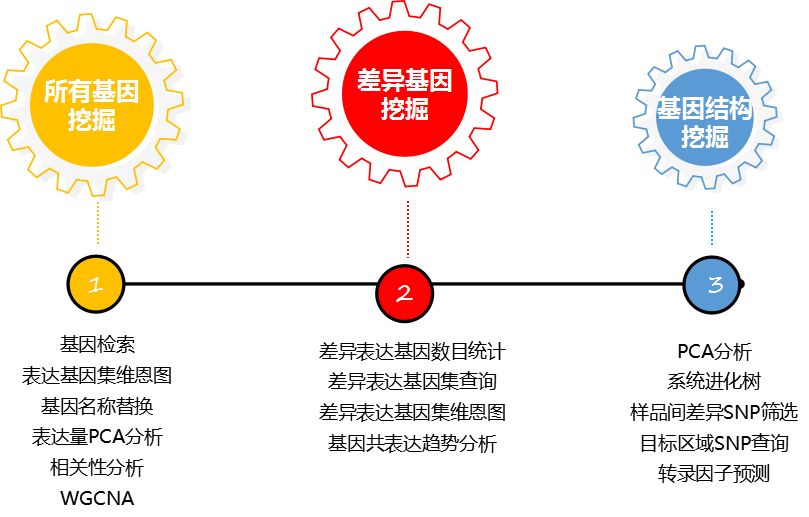
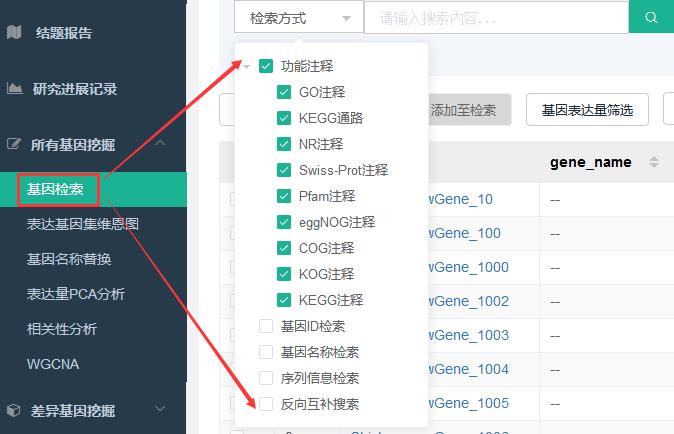
以“基因检索”功能为例,可通过基因ID、name、功能(8大数据库)、序列来检索目标基因/基因集,支持同时输入多个条目检索。
我们如何借助转录组分析APP实现经典数据挖掘思路呢?首先需要登录百迈客云(www.biocloud.net)——我的项目——进入相应项目【报告】。然后通过以下操作就可以啦:
01
数据挖掘之自定义筛选DEG
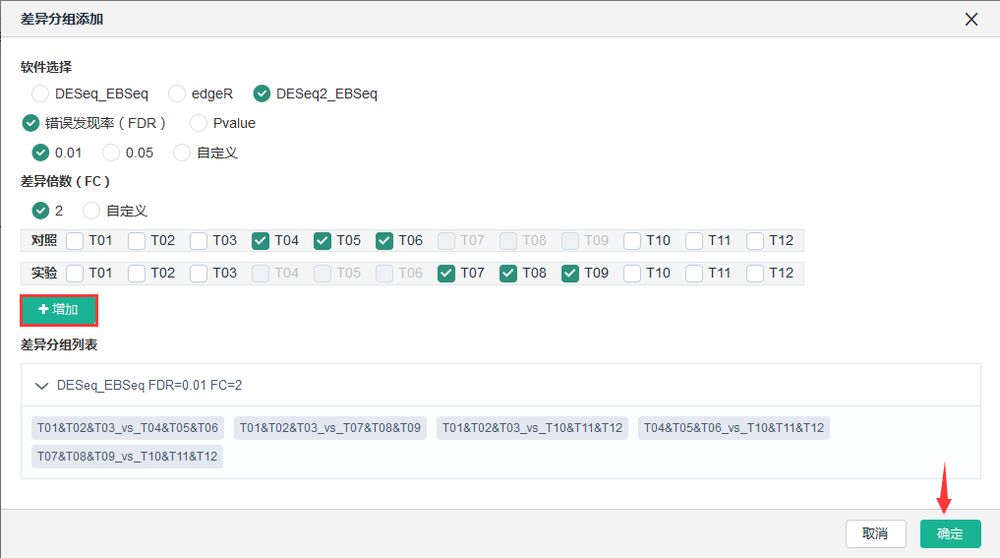
新增差异分组,调整FC(Fold Change)及FDR(False Discovery Rate,错误发现率)筛选差异表达显著的基因。
操作步骤:
差异基因挖掘——差异表达基因集查询——添加差异分组、自定义软件、自定义参数(FDR或p-value、FC)——【添加】——【确定】
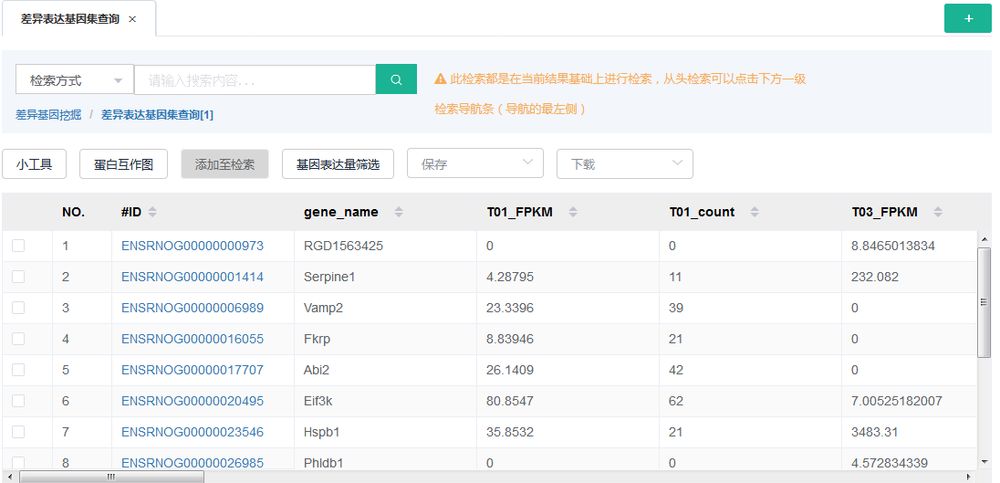
得到的结果表格中包含基因ID、name、表达量、FDR、FC、up/down、注释。此外,也可以筛选其中的“只上调/只下调”的DEG。
步骤
结果界面
左右滑动查看图片
说明:差异基因挖掘中提供了两个软件——DESeq1_EBSeq和DESeq2_EBSeq,DESeq和EBSeq分别用于有生物学、无生物学重复分析。DESeq2是DESeq的升级版,DESeq2在DESeq基础上升级成“shrinkage estimation”算法计算基因的count值。
02
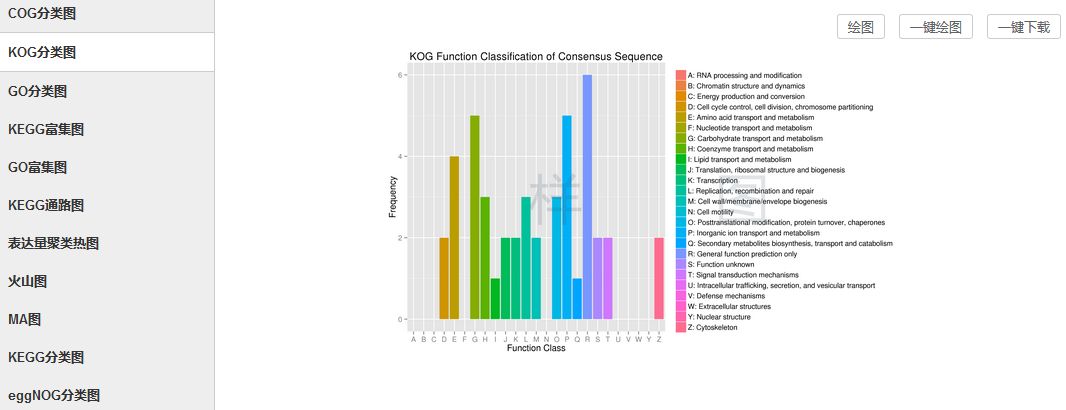
数据挖掘之DEG功能分析
对筛选到的基因进行功能注释与富集分析,一般是基于8大数据库进行注释,如GO、KEGG、COG、KOG等。
操作步骤:
点击筛选到的基因表格下方的“绘图”/“一键绘图”即可。
03
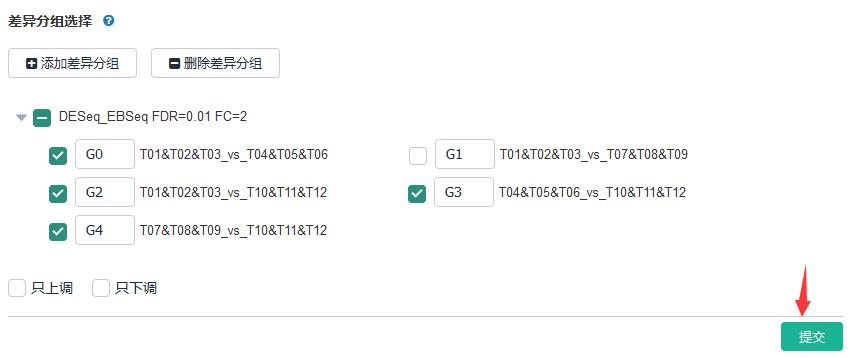
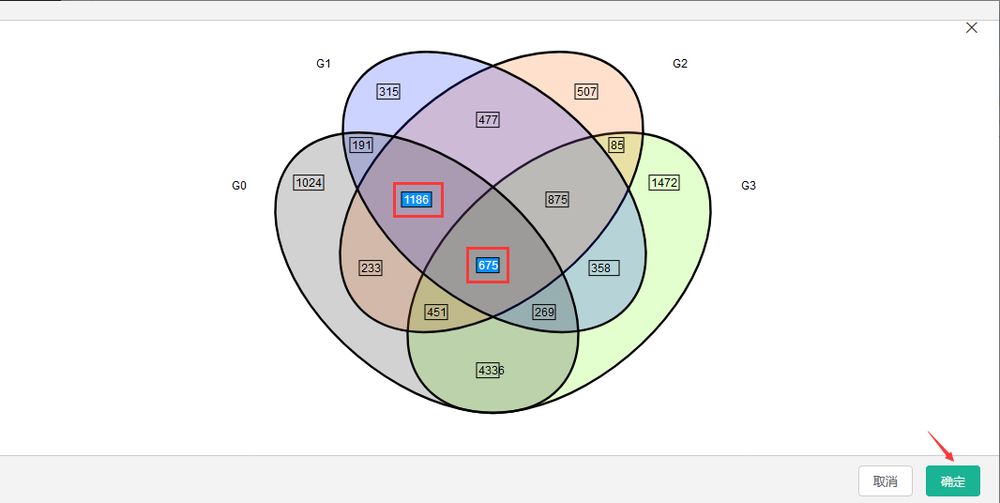
数据挖掘之组间DEG维恩图分析
筛选不同组合间共有及特异表达基因集合,通过GO分类、KEGG通路等分析来解析其特异或共有性状,阐明不同组合间异同的作用机理。
操作步骤:
差异基因挖掘——差异表达基因集维恩图——选择差异分组——【提交】
当然,也可以针对只上调或者只下调的基因绘制维恩图。此外,绘制出的维恩图可以实现在线的可视化交互,即可点击图中某个/某些基因集,便可得到对应基因的具体信息表格。
步骤

结果界面
交互
左右滑动查看图片
04
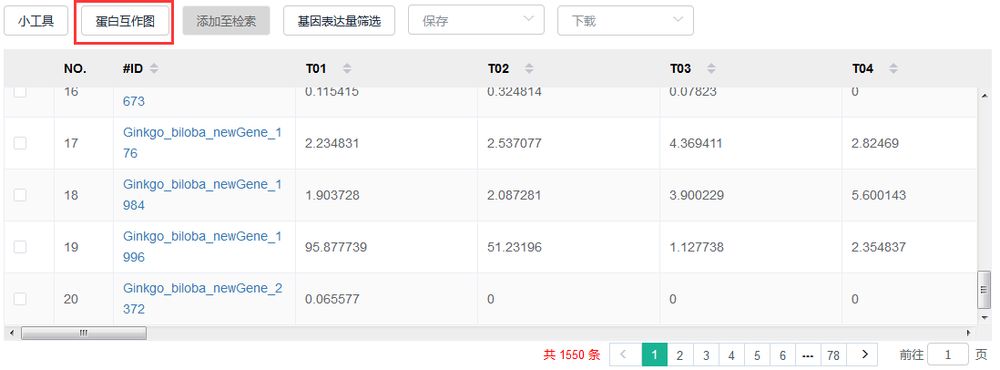
数据挖掘之DEG蛋白互作分析
基于STRING数据库,对筛选到的基因进行蛋白互作分析,绘制蛋白互作网络图。
操作步骤:
点击筛选到的基因表格上方的“蛋白互作图”即可。
步骤
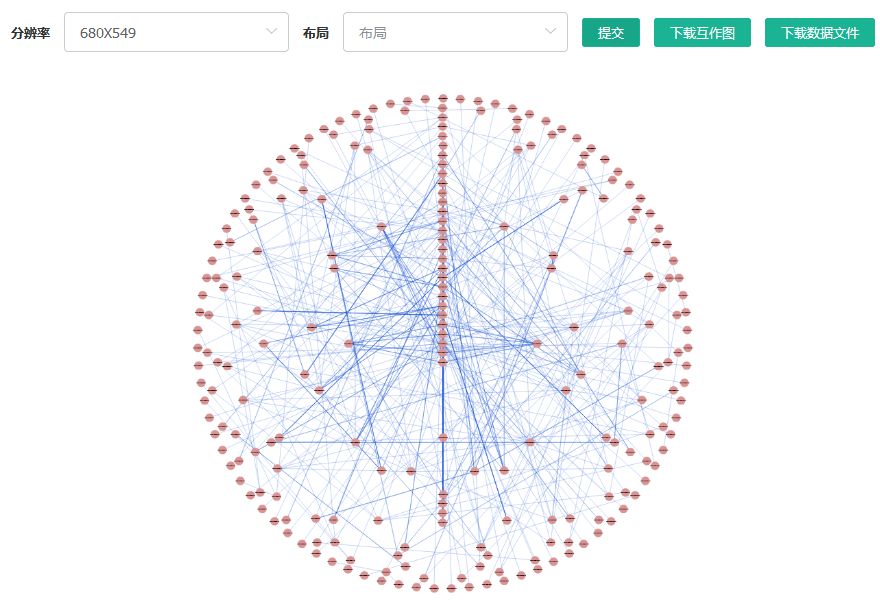
结果界面
左右滑动查看图片
05
数据挖掘之K均值聚类
对特定的DEG在特定样品中的表达趋势进行分析,得到表达趋势一致的基因集。
操作步骤:
差异基因挖掘——基因共表达趋势分析——“差异分组列表”中选择需要进行分析的DEG集——“样品选择”中选择需要进行分析的样品——设置“K值”(该值代表表达趋势类别数)——【提交】
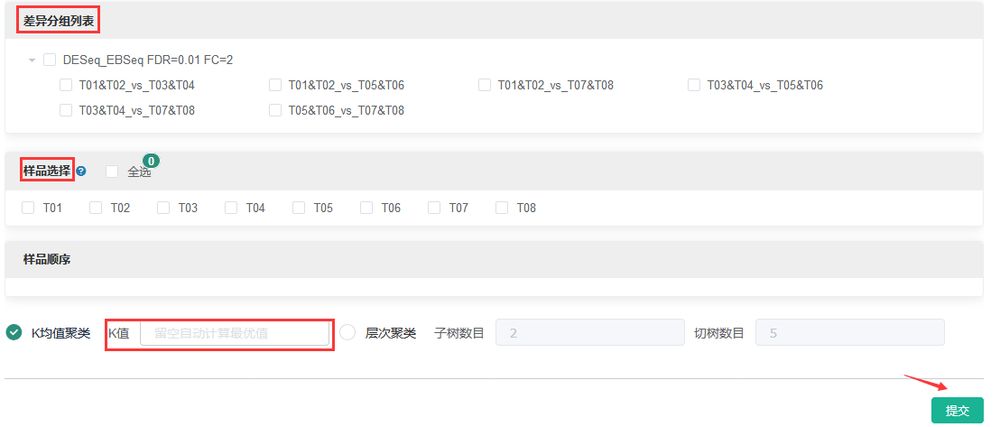
步骤
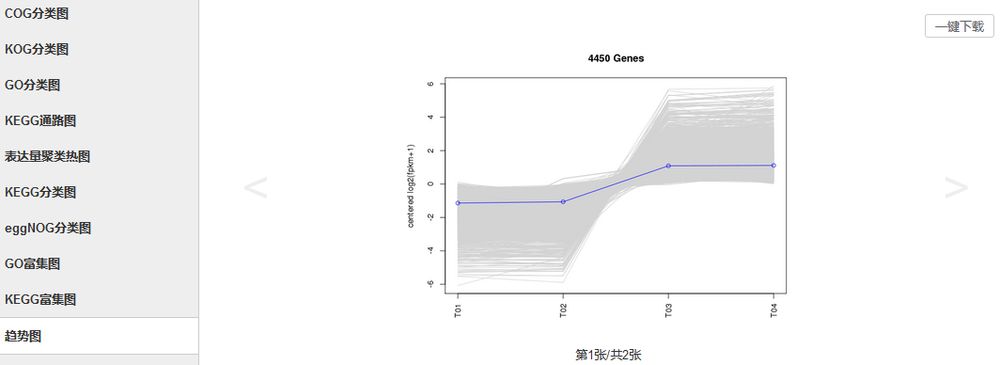
结果界面
左右滑动查看图片
06
数据挖掘之WGCNA
对特定的基因进行基因共表达网络分析,将基因分为不同的模块,探索模块与特定表型或疾病的关联关系,筛选关键基因集。
操作步骤:
所有基因挖掘——WGCNA——选择“质量性状”/“数量性状”分组——选择需要分析的样品,设置分组——若是数量性状分组,则需填写性状值——【确定】。
此外,表达阈值、模块相似度阈值、模块内最小基因数这几个参数支持自主设置与调整。
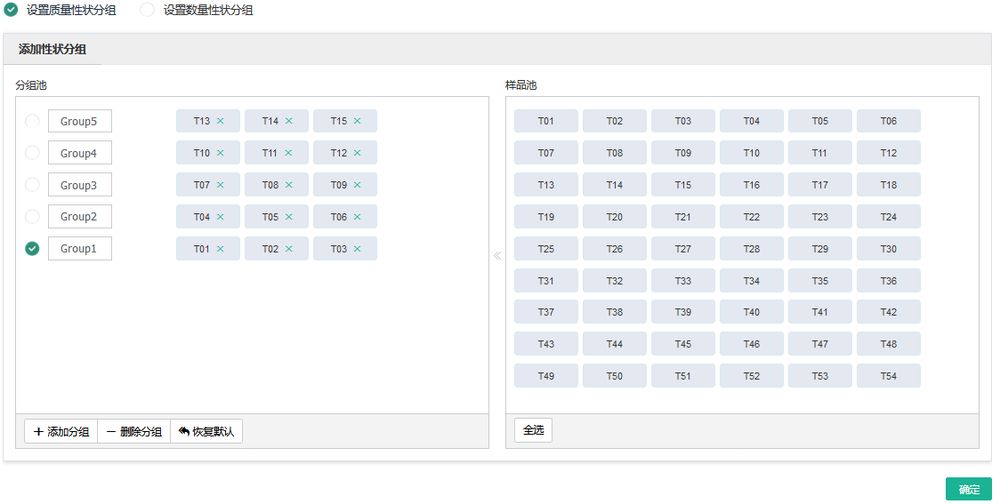
步骤
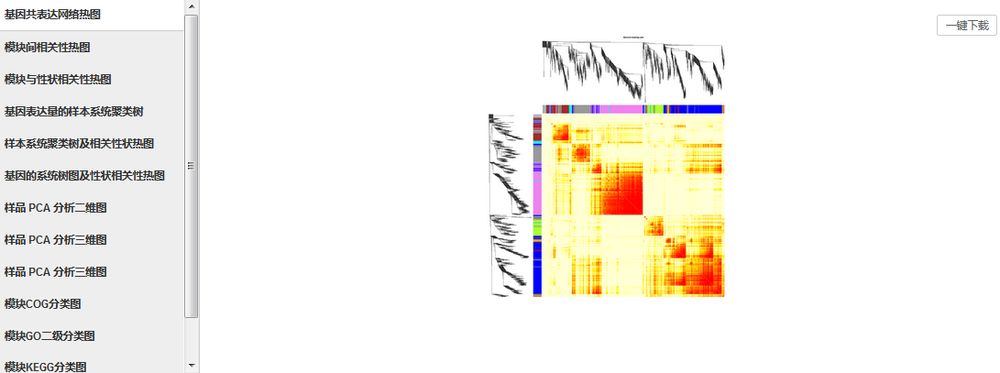
结果界面
左右滑动查看图片
是不是很easy呀

今天就介绍这么多,眼过百遍不如手过一遍,我们为大家提供了可以极速体验APP的项目数据,即便没有测序项目也可以体验哦,通过以下几步便可快速体验:
登录云平台——分析——医学/农学——转录组分析平台——运行极速体验数据——个性化分析
赶紧趁热去百迈客云平台试试吧。
最近转录组测序还有优惠活动哦,点击下方“阅读原文”了解详情。
以上是关于转录组测序1-测序原始数据说明的主要内容,如果未能解决你的问题,请参考以下文章