转录组测序中 gene 和transcript 的区别
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了转录组测序中 gene 和transcript 的区别相关的知识,希望对你有一定的参考价值。
参考技术A 没记错的话,一个gene可以有多个transcript(也就是isoform)本回答被提问者采纳非链特异性转录组测序
非链特异性转录组测序
转录组(transcriptome)在广义上是指细胞内全部转录产物的集合,狭义上是指细胞中所有转录本(mRNA)的集合。转录组测序通常来说是指依托于高通量测序平台对细胞中的mRNAs进行测序。经过信息的整理和分析,可在不同的样本中挖掘具有意义的差异表达基因,研究可变剪切,融合基因事件和在转录水平上进行性状定位等。转录组测序现已广泛的应用到科研基础研究,临床疾病研究和药物研发等领域。
常规转录组测序根据实验样品的不同可分为真核生物转录组测序和原核生物转录组测序;根据建库时的方法和研究目的不同可分为链特异性转录组建库和非链特异性转录组建库;根据测序物种有无参考基因组可以分为有参基因组转录组测序和无参基因组转录组测序。技术路线
有参考基因组的转录组测序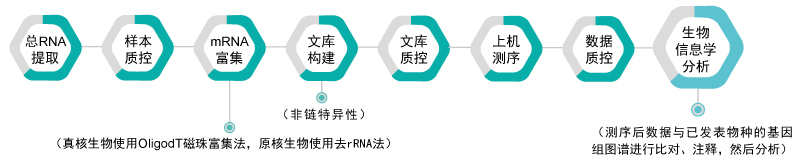
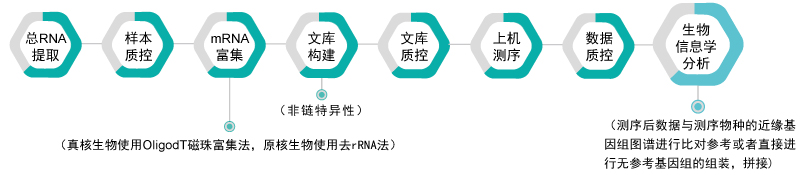
无参考基因组的转录组测序
技术优势

测序策略
| 测序策略 | PE100或PE150 |
| 建议数据量 |
真核生物:一般测序5-6G深度测序10G 原核生物:一般测序1G深度测序5-10 |
收样要求
| 组织 | 500mg新鲜植物样本,300mg新鲜动物样本注: 肿瘤组织优先选择RNAlater保存 |
| 细胞 | >5*106悬浮/贴壁细胞 |
| 血液 | >3ml全血/全血分离的有核细胞 |
| RNA总量 | >3 μg,浓度 >50ng/μl, RIN值>6.5 |
| 对于较难进行取材的医学类样品,可以适当降低样本标准。 | |
您可以得到的数据分析
有参非链特异性转录组生物信息学分析流程
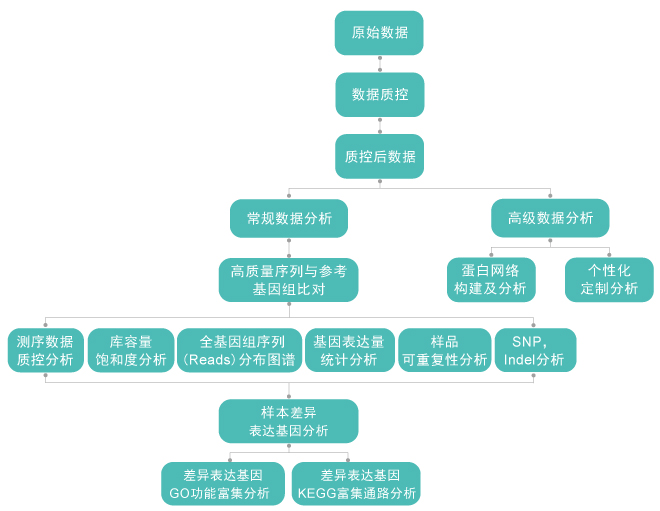
|
常规分析 |
高级分析 |
|
高通量序列与参考基因组比对 测序数据质控分析 基因表达量分析 基因的可变剪切分析(真核生物) 基因结构优化及新转录本预测 SNP、InDel等基因结构变异筛查 样本间基因差异表达分析 差异表达基因的KEGG和GO功能分析 |
差异基因的蛋白互作分析 个性化定制分析 |
无参(de novo)非链特异性转录组生物信息学分析流程
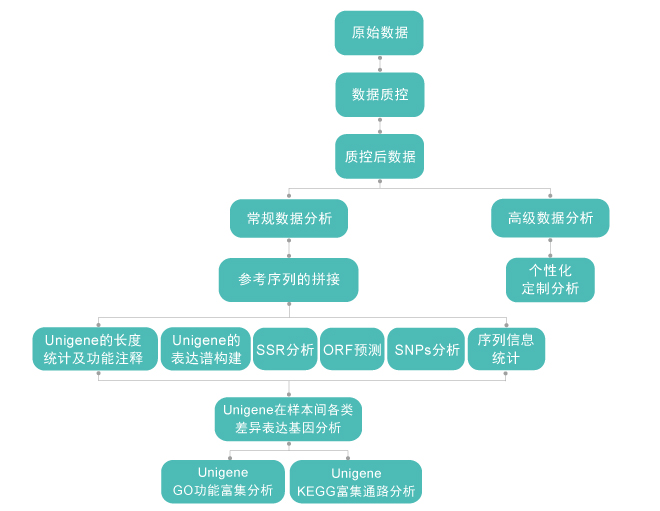
|
常规分析 |
高级分析 |
|
测序数据质控分析 参考序列拼接 Unigene的长度统计及功能注释 Unigene的表达谱构建 SSR分析 ORF预测 SNPs分析 序列信息统计 Unigene在样本之间各类差异基因表达 Unigene的KEGG和GO功能分析 |
差异基因的蛋白互作分析 个性化定制分析 |
案例解析
利用肿瘤血小板RNA-seq发现直接利用血液在分子通路层面上诊断
泛癌症、分辨不同癌症类型和鉴定癌症基因突变的方法
(有参转录组测序)
肿瘤相关的血小板(Tumor-educated platelets, TEPs)在全身或病灶范围上会根据肿瘤的生长情况而改变自身mRNA的表达。本研究对228个原发肿瘤或肿瘤转移患者和55个健康个体血液中的血小板细胞进行了转录组测序,欲验证肿瘤血小板中mRNA的变化是否能指导癌症的诊断和分类。测序后,通过序列比对,共鉴定出5,003个已知的血小板mRNA生物标记,例如B2M, PPBP, TMSB4X, PF4等,同时也鉴定出了一些非编码RNA(lncRNAs)。
癌症患者与健康个体相比,1,453种mRNA表达量提高,793种mRNA表达量下降。聚类分析结果显示癌症患者与健康个体的血小板细胞mRNA信息有较大差异。随后,课题组基于差异表达基因,利用不同算法对6种不同的癌症类型进行诊断和区分,结果显示可达到较好的癌症分辨效果。
本研究通过对血小板细胞进行转录组测序,发现了基于肿瘤相关血小板细胞中mRNA的信息对肿瘤进行早期诊断和癌种区分的方法。
Best, Myron G., et al. "RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics."Cancer cell(2015): 666-676.(Imapct factor:27.407)
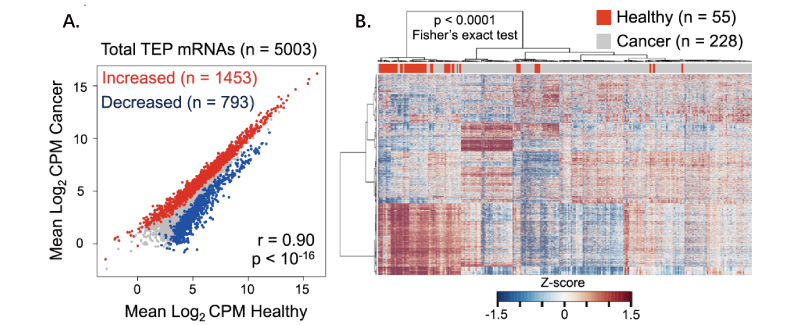
A. 测序后分析5,003个mRNA的上下调表达情况(红色代表上调基因,蓝色代表下调基因)
B.健康个体和肿瘤患者mRNA表达量差异热图(上方横条中红色代表健康个体,灰色代表肿瘤患者)
蝾螈不同组织de Novo转录组测序鉴定与肢臂重生相关mRNA与其编码的蛋白质
(无参转录组测序)
哺乳动物的肢体再生能力非常有限,蝾螈却可以重生自身肢臂。对蝾螈再生肢臂的机制的理解并不透彻,其中一个重要的原因是蝾螈基因组巨大且不完整。本研究对蝾螈16种不同组织(包括肢臂胚芽组织、睾丸、肌肉、卵巢等)的共42个样品进行转录组测序,并de novo拼接测序数据,绘制出了一个近乎于蝾螈全转录组的图谱 (完整率高达88%)。同时,通过对比分析肢臂再生组织和其他组织的mRNA测序数据和表达情况,并结合先前文献,鉴定出了在肢臂再生中特异性表达的转录本和其编码的蛋白;CIRBP和KAZALD1蛋白。本研究除完成高完整度的蝾螈全转录组图谱构建及注释外,还揭示了在蝾螈进行肢体重生中的重要基因,极大地填补了蝾螈肢体再生研究的空白,并为再生医学研究奠定了基础。
Bryant, Donald M., et al. "A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors."Cell reports(2017): 762-776.(Impact factor: 8.282)
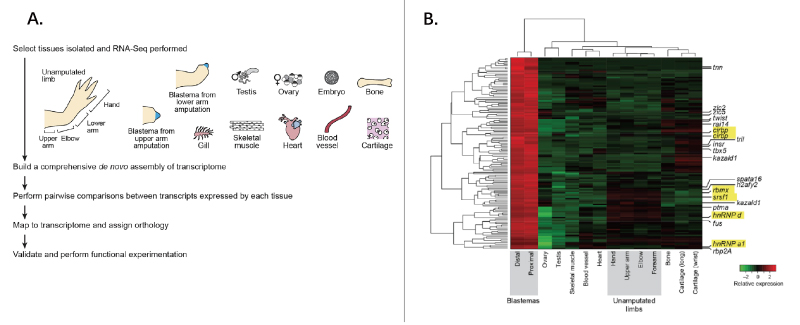
A. 针对蝾螈16种不同组织样本的转录组测序和de novo拼接流程
B. 159个在肢臂胚芽组织中富集的mRNA和在各种组织中的表达热图,标黄的基因代表预测为可能与肢臂再生相关的基因
研究趋势与研究热点
寻找研究对象的重要性状、表型或生物标志物;
构建研究对象在不同的处理条件下,或者不同表型研究对象的基因表达谱。寻找表达差异大的关键基因;
研究对象时序性或空间特异性变化的主控因素;
无参考基因组物种的基因表达谱构建;
研究近缘物种之间的进化差异等。
常见问题
在进行转录组测序后,生物信息学分析会展现出不同样品中差异较大或有特殊意义的转录本,为了对这些有意义的转录本进行验证和继续研究,qRT-PCR方法可通过对目标转录本的表达进行定量或定性验证。除此之外,还有Northern Blot,FISH(荧光原位杂交)和免疫荧光实验等方法也可用于测序结果验证。
转录组一般要测多大的数据量才合适?
转录组测序所需要的测序量要根据多个因素考量确定,这些因素包括了待测物种转录组大小,待测物种转录组中基因的数量和各类基因的丰度。不同的物种在以上因素的变化可能不同,因此为了保证数据分析结果的可靠性和准确性,我们建议客户一定要在测序之前对待测物种的转录组的大小进行评估。对于有参考基因组的物种,可以根究先前的经验,分析基因组信息、编码基因个数、碱基数及丰度进行预测评估。大部分物种的测序量以6-8G为佳。而多倍体植物,例如八倍体小麦这类基因组较大且复杂的物种来说,我们推荐适当增加测序量至10-12G。
转录组测序的数据质量影响因素主要有哪些?
转录组测序的质量可能会受到以下因素的影响:(1)RNA严重的降解可能会影响建库与测序的质量 (2)过低的RNA起始量可能会影响建库与测序质量,可以通过适当地增加PCR循环数来解决 (3)转录组中基因丰度的较大差异可能会影响基因检测的分辨率,高丰度的基因可能会掩盖低丰度表达的基因。以上是关于转录组测序中 gene 和transcript 的区别的主要内容,如果未能解决你的问题,请参考以下文章