GSEA背后的统计学原理
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GSEA背后的统计学原理相关的知识,希望对你有一定的参考价值。
参考技术A所以GSEA目的是找到你筛选出来的基因,是否在你感兴趣功能的基因集里面有富集,那么GSEA符合什么样的一个统计学原理呢?
也就是说经过差异表达筛选出来的Gene List 首先根据P-value(Rank Metric)进行排名,Rank Metric 小的在上面,大的在下面
富集分数:
Equal weights: The ‘classic’ Kolmogorov-Smirnov statistic:
GSEA软件计算最终的富集得分即为上图对应于ecdf图之间最大的垂直距离(绿色虚线),也称为峰值
Kolmogorov-Smirnov拟合优度检验
单样本Kolmogorov-Smirnov拟合优度检验
该检验的目的是检测实际样本的分布是否与理论分布是否相契合
基本原理 : 将理论分布下的累计频数分布与观测到的累计频数分布相比较,找出它们之间的最大差异点(即图中绿色的线所在的点) , 并参照抽样分布,判断最大差异是否来自偶然
其中:
双样本Kolmogorov-Smirnov拟合优度检验
表型(功能)置换
P-value 计算
标准化的富集得分:考虑基因组大小
翻译自:
Gene Set Enrichment Analysis
GSEA - Gene set enrichment analysis 基因集富集分析原理与应用
RNA-seq是利器,大部分做实验的老板手下都有大量转录组数据,所以RNA-seq的分析需求应该是很大的(大部分的生信从业人员应该都差不多要沾边吧)。
普通的转录组套路并不多,差异表达基因、富集分析、WGCNA network以及一些没卵用的花式分析。DEG分析是基础,up and down,做个富集,了解一下处理后到底是什么通路被改变了;WGCNA主要就是根据相关性来找出一些co-express的gene module。
单细胞的转录组的玩法就比较多了,可以理解为超多样本的普通转录组,普通转录组的分析基本都可以用,但单细胞更侧重于两个主题:clustering和pseudotime。
RNA-seq离不开富集分析的本质原因就是因为它是超高通量的,老板肯花钱做RNA-seq自然就是想做数据挖掘,而不是focus on one gene;3万个基因你怎么分析?就算你用WGCNA得到了很多in silico的gene module,so what?生物老板不懂计算机,你给他们一大堆冰冷的gene有什么用,所以一些超级经典的数据库就出现了,GO、KEGG等,3万多基因我都根据现有知识给你做了定义定位分类,这样老板就知道,原来我敲除了A gene会导致B通路下调啊,这样我就可以接着讲我的生物学故事了。真找到老板感兴趣的通路,老板故事讲好了,你就可以午饭多加个鸡腿了。
从基因表达或者基因集到富集通路的分析过程就是GSEA了。
GSEA是一个软件的名字,就是基因集富集分析的意思,但它其实是特指某一种基因集富集分析;在GSEA之前就已经有了普通的基因集富集分析方法;两者之间的目的是一样的,但是原理有天壤之别。
先列一个富集分析的常见工具表:
- DAVID - 很多CNS里都会见到,后来被人发现它根本就不更新数据库
- WEGO - 华大在用的,Y叔还和华大有口水战
- clusterProfiler - Y叔精品
- webgestalt - 网页版的,也有R包,之前我在用
- ClueGO
- goProfiles
- MeV
- TEASE
GSEA单独列出,GSEA - Broad Institute,broad出品必属精品。
一些常用的数据库:
- GO
- KEGG
- DO - Disease Ontology
- Molecular Signatures Database (MSigDB)
- webgestalt里可以看到更多的数据库
普通富集分析的原理比较简单,可以直接看clusterprofiler的文章,一下是原文截取:
The clusterProfiler package depends on the Bioconductor annotation data GO.db and KEGG.db to obtain the maps of the entire GO and KEGG corpus. Bioconductor annotation packages org.Hs.eg.db, org.Mm.eg.db, and org.Sc.sgd.db were imported for genome-wide annotation of mapping Entrez gene identifiers or ORF identifiers for humans, mice, and yeast, respectively.
The clusterProfiler package offers a gene classification method, namely groupGO, to classify genes based on their projection at a specific level of the GO corpus, and provides functions, enrichGO and enrichKEGG, to calculate enrichment test for GO terms and KEGG pathways based on hypergeometric distribution. To prevent high false discovery rate (FDR) in multiple testing, q-values (Storey, 2002) are also estimated for FDR control. Furthermore, clusterProfiler supplies a function, compareCluster, to automatically calculate enriched functional categories of each gene clusters and provides several methods for visualization.
稍微梳理一下:首先该工具依赖一些数据库GO和KEGG,另外需要一些基因名转换的数据库;然后groupGO就是根据GO term来把我们的基因集分类,然后enrichGO就是来做统计检验(超几何分布),判断这个基因集是不是随机抽取的;然后就是一些统计的修正了,FDR和q-value。
以下列几个我经常会碰到的问题(大部分都被Y叔解决了):
- 数据库冗余,大部分条目都是重复的,解决:use simplify to remove redundancy of enriched GO terms
- 一些太general的条目出现了,需要过滤,解决:test GO at sepcific level
- 一些term的名字太长,解决:看最新的公众号,2019年02月13日
- 有些基因名字不匹配,我是直接用的基因symbol的
能做好普通富集分析已经不错了,但是你看高分paper,发现更多大佬都在用一个叫GSEA的分析方法来做富集分析。以下就是这种分析的典型结果图:
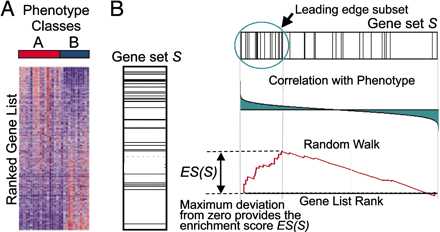
以前我死活看不懂这张图(因为大部分教程都不说人话),后来偶然看到一篇教程,真的是通俗易懂,对着上面的图一看,理解了就再也忘不了。
附教程链接:GSEA分析结果详细解读
普通富集分析的致命缺点:
- 已经选出了DEGs,需要主观的过滤
- 在统计检验的时候不考虑基因的表达情况
- 一些微弱的却具有效力的基因集被过滤掉了
这就是为什么有些老板结果看多了,就自然理解出了普通富集分析的缺点,我给的基因本来就是偏向于某些通路的(比如说大脑发育的样本),那我注释出来的结果自然就有很大一批大脑发育的通路,这是必然的confounder。这就极大地削弱了富集分析结果的准确性!!!在这里我敢大胆的说,大部分paper里的普通富集分析结果都是为赋新词强说愁,为了讲故事而讲故事,根本不具备任何的科学性。
broad的人估计是灌水太多,不忍再残害科学界,才花了大力气打造了一个颇具科学性的基因富集分析工具GSEA。
待续~
以上是关于GSEA背后的统计学原理的主要内容,如果未能解决你的问题,请参考以下文章