基于MS强度或计数的数据依赖法非标记定量蛋白质组学的蛋白质互作分析
Posted xixilichuanshuo
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了基于MS强度或计数的数据依赖法非标记定量蛋白质组学的蛋白质互作分析相关的知识,希望对你有一定的参考价值。
本期接着上期内容,继续给大家介绍基于MS强度或计数的数据依赖法非标记定量蛋白质组学的蛋白质互作分析。
通过对MS1扫描中检测到的信号强度的面积进行测量可以获得定量信息:目标物质的质量可从时间域中提取,从而生成提取离子色谱图(XIC),可用于整合目标化合物的LC洗脱峰。这一方法类似于处理SILAC数据的方法,它可确定同位素代码对的谱峰强度。这些基于MS1的非标记方法和同位素标记方法的主要区别在于,前者必须通过结合保留时间标记或重新调整保留时间数据的算法来密切管理LC中的保留时间。使用LC系统也可以进行调整,LC系统在洗脱方面具有很好的再现性。为了准确定义LC洗脱峰,仪器的扫描速率也很重要,因为测量不足会对面积测定造成很大影响(图2)。尽管图2未明确标出,但通过LC峰的数据点的数量减少会使得峰顶的数量明显减少并影响面积测定。在鸟枪法测序流程中,定量的质量也可能有问题,其中MS1扫描的频率受到进行碎片化的离子的选择和每个前体离子测序所花时间的影响。新一代仪器能够收集大量MS/MS信号,并在LC洗脱峰上保持一致的点数,因此能提供可靠的基于MS1的定量。
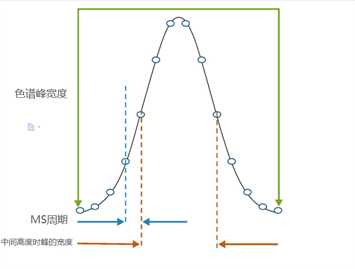
图2. 用于确定峰形状的点数对定量重现性至关重要。周期和峰宽度之间的平衡是确定色谱峰形状的关键。用于定义LC峰形状的建议最小点数是8个点,这个数量能很好地预估峰值。周期是累积时间和监测/碎片离子数量之间的一个平衡。在鸟枪法蛋白质组学中,LC峰很少被取样,而SRM分析中的这种取样较多。因此,定义峰值不仅需要有大量的点,这些点之间得是等距离也同样重要。
一般来说,基于MS1的量化与鉴定同时进行,仅可对已经经过鉴定的种类进行量化。但是也有例外:例如,MS1定量在代谢组学中广泛使用,且不需要用MS/MS测量来鉴定化合物。在这种情况下,依次排列样品的MS1色谱峰,并描出区域的轮廓。在蛋白质组学中,可以采用类似的数据简化过程,包括按不同样本的质量和保留时间对齐MS1数据,然后进行统计处理,以鉴定出那些在生物学上有显著差异的。该数据处理方案通过预测肽保留时间的方法得以优化,允许在没有MS/MS的情况下进行随后的化合物鉴定。在蛋白质互作分析的背景下,还有一种中间策略,混合参考样本的鉴定结果可用于瞄准MS1数据提取和随后的量化工作。这种混合方法支持以半瞄准方式从样本中提取数据,并且在许多情况下不需要进行复杂的LC-MS轨迹对齐这个步骤。
本文由百泰派克生物科技整理编辑。未经许可,不得转载。
百泰派克生物科技专注于基于质谱的蛋白质组学服务,结合亲和纯化与非标记定量蛋白质组Label-free、SILAC或SWATH定量技术服务,提供一系列的蛋白质组研究策略,灵敏度高、重复性好,非常适合蛋白质相互作用的研究。
文献参考:Stephen Tate, Brett Larsen, Ron Bonner, Anne-Claude Gingras, Label-free quantitative proteomics trends for protein-protein interactions. Journal of Proteomics, 2013.
以上是关于基于MS强度或计数的数据依赖法非标记定量蛋白质组学的蛋白质互作分析的主要内容,如果未能解决你的问题,请参考以下文章
无标定量|有标定量|谱图计数|XIC|AMT数据库|RT对对齐|母离子|子离子|SILVER|SRM|iBAQ|APEX|差异蛋白筛选|MaxQuant|PANDA|C-HPP