非靶代谢组学数据预处理XCMS+IPO
Posted 修罗神天道
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了非靶代谢组学数据预处理XCMS+IPO相关的知识,希望对你有一定的参考价值。
质谱可以用来测量样品中带电分子(离子)的丰度(即一个特定的质荷比 m/z的离子计数)。
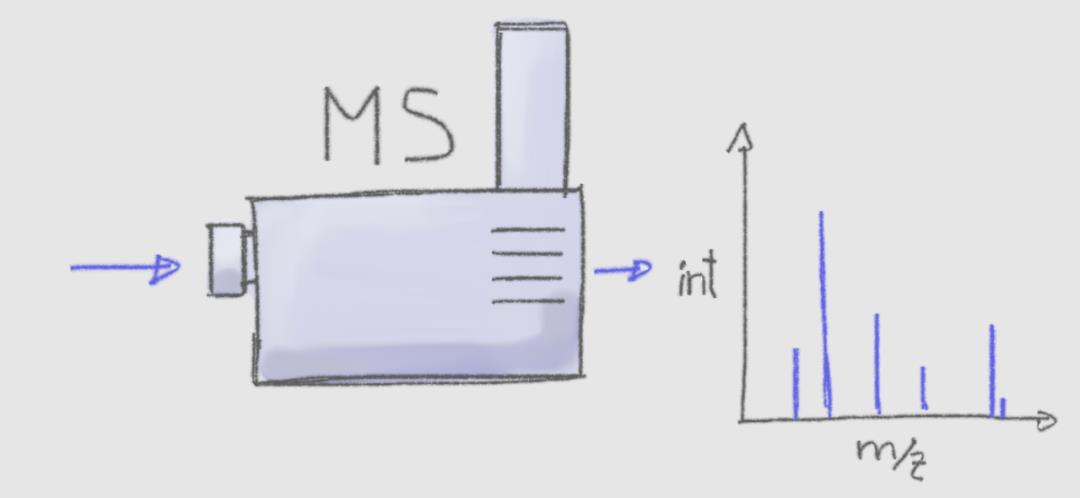
但是,当许多离子具有相同或非常相似的 m/z,光凭借质谱是很难或不可能进行区分的。因此,MS 经常与其他分离技术相结合,基于除 m/z以外的其他性质来分离分析物。比如与气相色谱法(GC)或液相色谱(LC)联用。
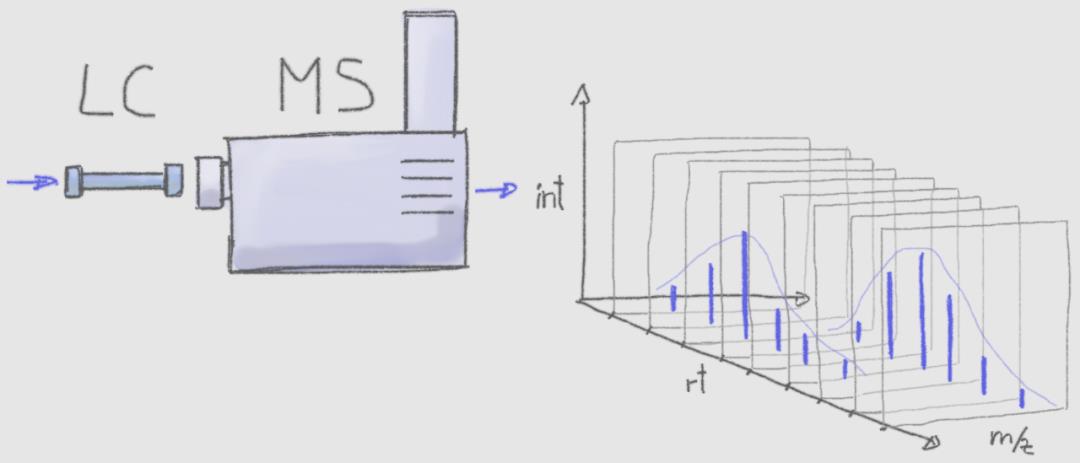
这些使用LC/GC-MS联用产生的数据,需要经过一系列的预处理,才能得到一个定量的表格数据。
开源软件中,XCMS是LC-MS数据前处理引用率最高的软件,在GC-MS中也很常用(第2)。但是由于数据的复杂性,一般来说,XCMS的默认参数通常都不是最优参数。因此,需要根据数据情况进行手动查看数据,来调整参数,又或者使用IPO这个R包来根据数据集情况对参数进行调优。
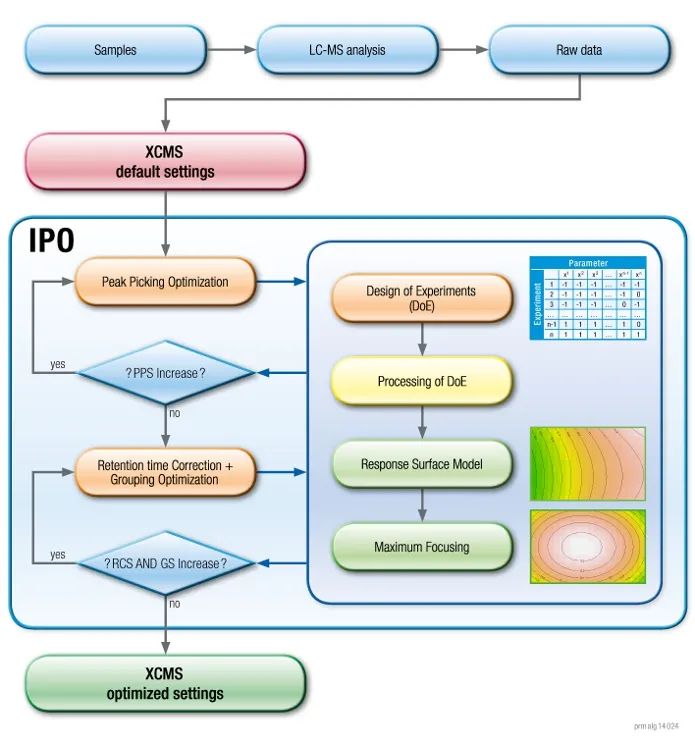
IPO(Isotopologue Parameter Optimization, “同位素参数优化”) 使用稳定的天然13C同位素来计算peak picking得分。保留时间校正是通过最小化特征内的相对保留时间差异来优化的,而grouping的参数是通过一次混合样品的injection中,只有1个峰的feature的数量最大化来优化的。下边是IPO参数调优的一个流程图。
软件准备
BiocManager::install(c("xcms","IPO"),update = FALSE, ask = FALSE)
BiocManager::install("faahKO")
调用R包
library(xcms)
library(IPO)
library(faahKO)
GC/LC-MS数据前处理
数据前处理包含3个步骤
-
chromatographic peak detection:色谱峰检测
-
alignment (also called retention time correction) :峰对齐
-
correspondence (also called peak grouping):峰分组
色谱峰检测
Chromatographic peak detection aims to identify peaks along the retention time axis that represent the signal from individual compounds’ ions.
简单来说就是,卡一个m/z范围,这个m/z范围理论上能代表某单个成分离子,检测这个离子沿着RT方向形成的峰。如下图的蓝色曲线。一条代表一个峰。
色谱峰检测的算法
findChromPeaks 函数,可以使用相应参数对象进行配置算法
| 算法 | 参数对象 | 说明 |
|---|---|---|
matchedFilter | MatchedFilterParam | 使用的是原始XCMS文章中描述的峰值检测算法 (Smith et al. 2006) |
CentWave | CentWaveParam | continuous wavelet transformation (CWT)-based peak detection(连续小波变换),可以检测出具有不同保留时间宽度的近邻和部分重叠峰(Tautenhahn, Böttcher, and Neumann 2008). |
massifquant | MassifquantParam | 基于卡尔曼滤波器的峰值检测(Kalman filter-based peak detection) (Conley et al. 2014). |
centWaveWithPredIsoROIs | CentWavePredIsoParam | Two-step centWave peak detection considering also isotopes |
MSW | MSWParam | Single-spectrum non-chromatography MS data peak detection(用于直接进样数据) |
其中,IPO支持的参数优化的算法:
-
centWave -
matchedFilter
下边用demo数据进行演示用法
cdfs <- dir(system.file("cdf", package = "faahKO"), full.names = TRUE,
recursive = TRUE)[c(1, 2, 5, 6, 7, 8, 11, 12)]
> cdfs
[1] "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/KO/ko15.CDF" "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/KO/ko16.CDF"
[3] "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/KO/ko21.CDF" "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/KO/ko22.CDF"
[5] "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/WT/wt15.CDF" "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/WT/wt16.CDF"
[7] "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/WT/wt21.CDF" "/Users/wangqingzhong/Library/R/3.6/library/faahKO/cdf/WT/wt22.CDF"
使用IPO找到最优参数:以matchedFilter算法为例。
# getDefaultXcmsSetStartingParams:获取起始参数。
peakpickingParameters <- getDefaultXcmsSetStartingParams('matchedFilter')
# 这个起始参数你可以修改,也可以不改。这里按照教程里来,还是进行修改
#setting levels for step to 0.2 and 0.3 (hence 0.25 is the center point)
peakpickingParameters$step <- c(0.2, 0.3)
peakpickingParameters$fwhm <- c(40, 50)
#对于设置的参数如果是单个数值,而不是2个数值,则认为这个参数不需要调整。
peakpickingParameters$steps <- 2
# 另外,没有必要使用所有数据进行参数调优,挑选其中的代表数据集即可。比如QC样品。这里是选择了前2个样品来调整参数。
time.xcmsSet <- system.time({ # measuring time
resultPeakpicking <-
optimizeXcmsSet(files = cdfs[1:2],
params = peakpickingParameters,
nSlaves = 1,
subdir = NULL,
plot = TRUE)
})
由于IPO使用的是XCMS比较老的函数xcmsSet,有一些参数的名称是有修改的。另外,部分参数是设定的2个数值(向量)作为一个参数,而得到的最优参数则是单个存储的,也需要进行一定的修改。
P.S.如果你还是继续使用老的函数xcmsSet来分析数据的话,在所有步骤的参数调优结束之后,可以使用writeRScript函数来获取分析脚本(内含最优化后的参数)。
bestparam <- resultPeakpicking$best_settings$parameters
paramPeakDetec <- MatchedFilterParam(
binSize = bestparam$step,
impute = "none",
baseValue = numeric(),
distance = numeric(),
fwhm = bestparam$fwhm ,
sigma = bestparam$sigma,
max = bestparam$max,
snthresh = bestparam$snthresh,
steps = bestparam$steps,
mzdiff = bestparam$mzdiff,
index = bestparam$index
)
此外,将这一步的结果存储起来,方便下一步的参数调优。
optimizedXcmsSetObject <- resultPeakpicking$best_settings$xset
使用最优参数对数据进行峰检测:
# 读取数据,mode = "onDisk"表示数据存在硬盘而非读入内存。这时候,内存大小的限制就会小一点,但是对IO的要求就会高一点。
raw_data <- readMSData(files=filePaths,
mode = "onDisk")
# 使用最优参数对数据进行峰检测
xdata <- findChromPeaks(raw_data, param = paramPeakDetec)
对齐
对齐(alignment)也称为保留时间校正。
色谱柱的保留时间漂移会比较严重。导致在不同样品之间的保留时间发生变化。峰对齐步骤就是为了调整样品之间的保留时间的漂移。
下图是base peak chromatograms(BPC)在保留时间校正前后得到的图。
所谓base peak chromatograms就是在每个RT挑选强度最大的峰作为这个RT的代表。
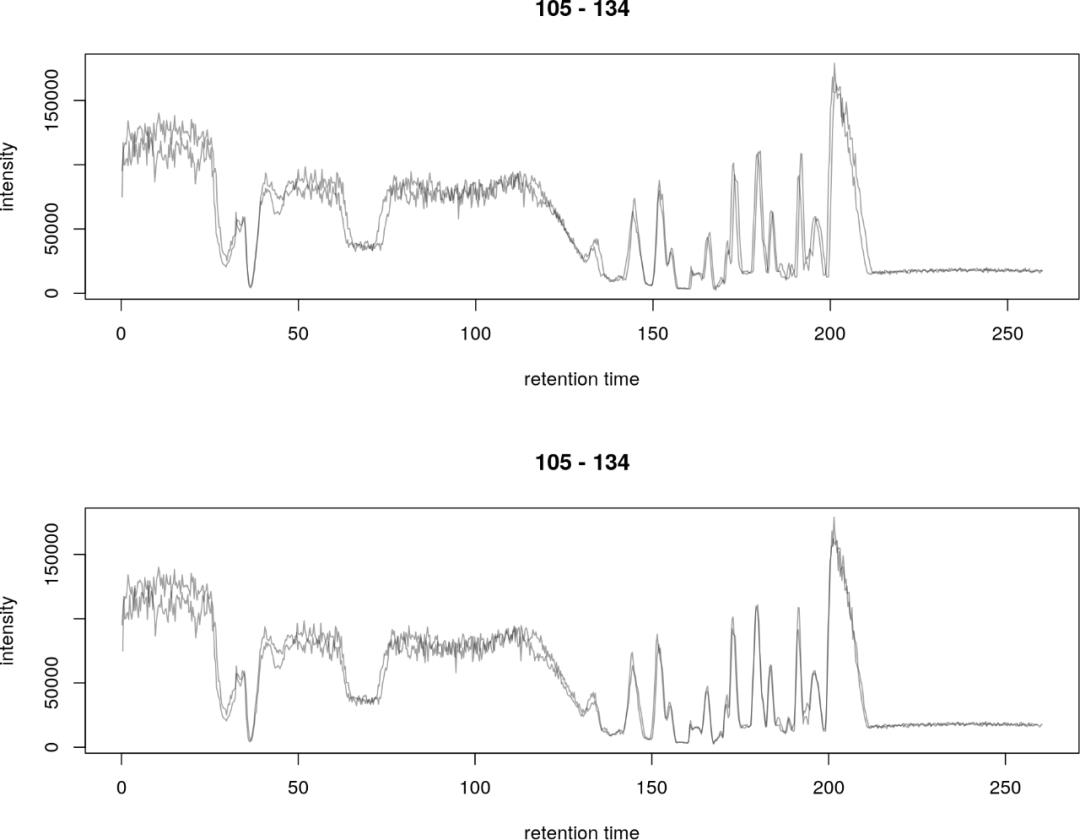
在XCMS里,使用adjustRtime函数进行峰对齐操作。
adjustRtime除了计算各谱图(spectrum)的调整保留时间(adjusted retention times)外,也对已鉴定的色谱峰(identified chromatographic peaks)的保留时间进行调整。
对齐算法
-
peakGroups(Smith et al. 2006),该方法根据hook peaks(钩峰)的保留时间对样品进行对齐,大多数样品中都应存在这些峰,由于它们被认为是来自同一离子种类的信号,可以用来估计样品之间的保留时间漂移。在对齐之前,我们必须对样本中的峰值进行分组,这是通过peakDensity correspondence分析方法完成的。 -
obiwarp(Prince and Marcotte 2006) ,manual里没对这个算法作过多说明,我也没有去找原始paper来研究。
两种算法都可以只使用其中一部分数据来做峰对齐(Subset-based alignment):
基于部分样品 (Subset-based alignment)
-
subset参数(PeakGroupsParamorObiwarpParam) -
subsetAdjust参数,allows to specify the method by which the left-out samples will be adjusted.-
举例:同上,B1将根据子集(QC)样品 A1和 A2之间的调整保留时间的平均值进行调整。而B3,B4由于其后没有QC样品,因此这两个样品只根据A2来调整。
-
请注意,加权平均数是用来计算调整后的保留时间平均值的,它使用非子集样本索引与子集样本索引差值的倒数作为权重。因此,像A1、B1、B2、A2这样的顺序,那么在对B1进行调整时,A1保留时间将比A2得到更大的权重,从而使其调整后的保留时间更接近A1而不是A2。
-
举例:A1, B1, B2, A2, B3, B4 ,如果A为QC,B为样品,使用
subset = c(1, 4)andsubsetAdjust = "previous",则A样品先跟彼此对齐,而其他没有被挑选出来的样品B1和B2会与A1对齐,B3,B4会与A2对齐。 -
subsetAdjust = "previous"
-
subsetAdjust = "average": 根据前一个和后一个子集样本的对齐结果的插值,调整非子集样本的保留时间。 -
两种情况都需要对样品进行有意义/正确的排序(例如,按进样顺序排序)。
-
请注意,对于任何subset-alignment,所有参数(例如minFraction)都是相对于子集的,而不是完整的实验!
-
而IPO支持的RT校正的算法:obiwarp 。
在IPO里是将峰对齐和峰分组放一起的。因此我们先介绍完峰分组再跑代码。
Correspondence
这一个步骤是将不同样品中的同一离子的色谱峰分到各自可以表征该代谢物的峰组(peak group), 也就是feature,从而得到feature的含量。
在XCMS里是使用groupChromPeaks函数来实现这一个步骤的。
算法
-
peak density (Smith et al. 2006)
-
nearest (Katajamaa, Miettinen, and Oresic 2006)
-
mzClust: high resolution peak grouping for single spectra (direct infusion) MS data.
其中IPO支持的仅支持 density。
# 同样先初始化参数。retcorGroupParameters <- getDefaultRetGroupStartingParams()resultRetcorGroup <- optimizeRetGroup(xset = optimizedXcmsSetObject, params = retcorGroupParameters, nSlaves = 1, subdir = NULL, plot = TRUE)
同样,对参数进行调整为新函数可以接受的样式。
使用最优参数对数据进行峰对齐和峰grouping:
xdata <- adjustRtime(xdata, param = paramAdj)
xdata <- groupChromPeaks(xdata, param = paramGroup)
在使用peak density 算法进行峰分组的时候,bw(bandwidth)参数特别需要注意。
这是bw为默认参数得到的峰group的结果。
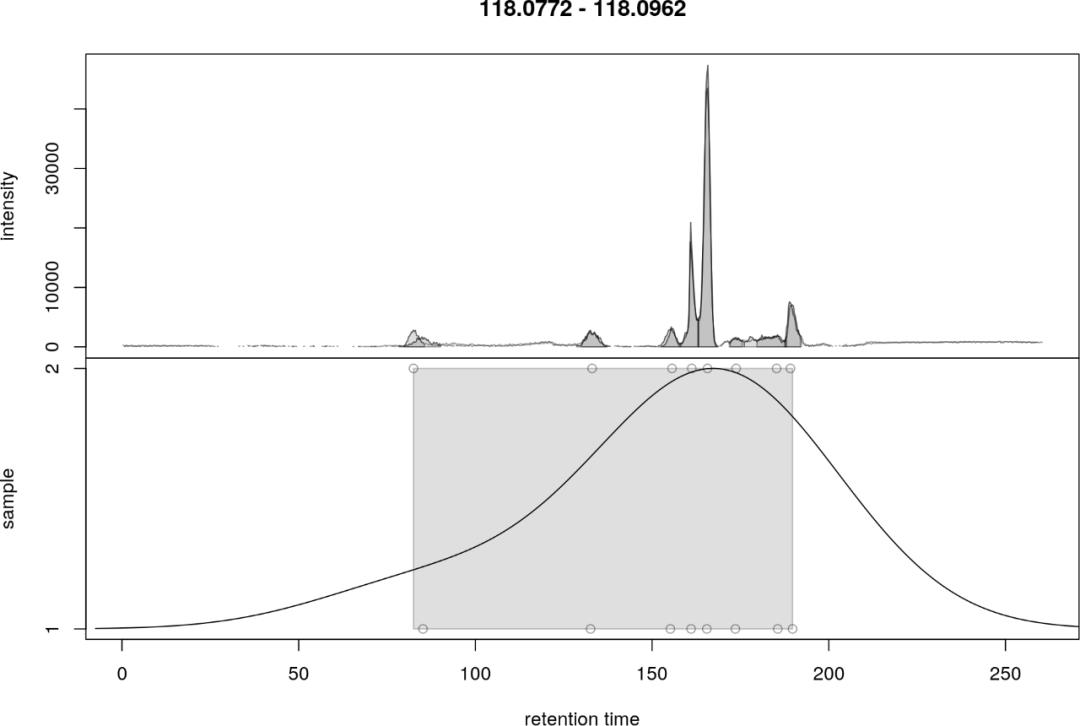
-
upper panel:色谱峰图
-
lower panel:沿RT轴分布的峰 (x-axis) ,y轴表示样品的数量,一个圆圈表示一个样品。黑色实现表示这些样品沿x轴的分布。
-
峰组 (features) 为灰色长方形. The peak density correspondence method groups all chromatographic peaks under the same density peak into a feature.
这是调整bw为1.8后的结果。具体怎么得到这个图,参考xcms的文档Metabolomics data pre-processing using xcms
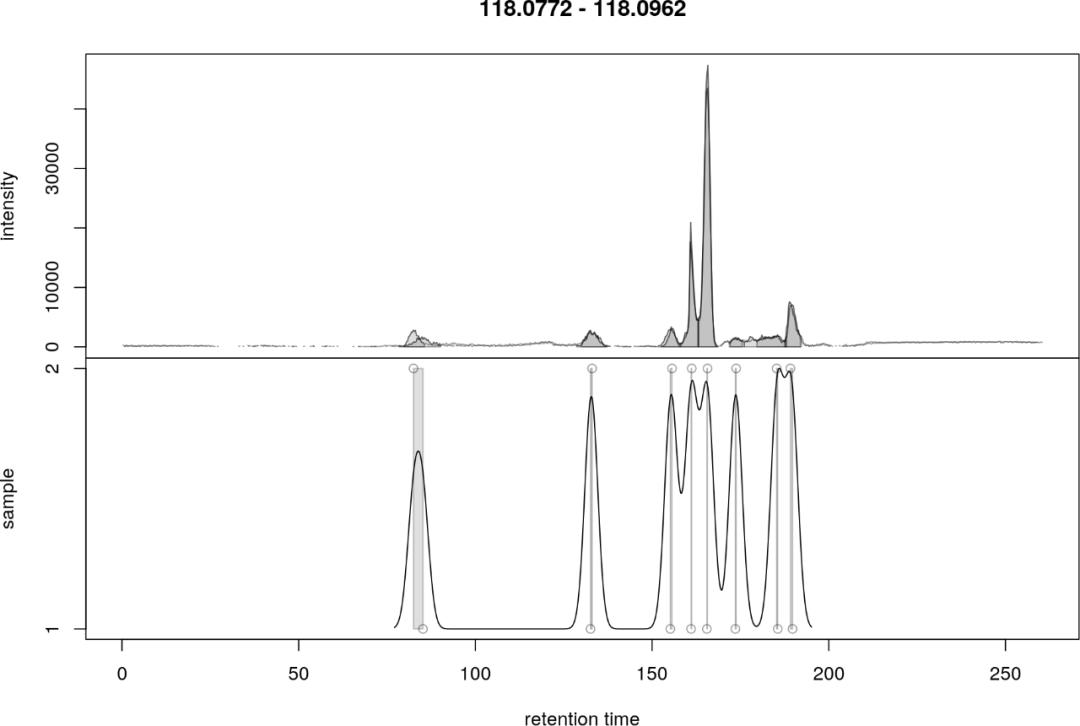
峰补齐(gap filling)
使用fillChromPeaks函数。
虽然大多数features的都有了丰度,但也会有一些NA。
如果未将色谱峰分配给feature,则可能会出现此类NA,原因可能是在各自的区域没有峰信号;也可能是有信号,但峰检测算法未能检测出色谱峰(如信号过低或噪声过大)。
head(featureValues(xdata, value = "into"))## ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF## FT001 NA 506848.9 NA 169955.6 NA NA NA## FT002 1924712.0 1757151.0 1383416.7 1180288.2 2129885.1 1634342.0 1623589.2## FT003 213659.3 289500.7 NA 178285.7 253825.6 241844.4 240606.0## FT004 349011.5 451863.7 343897.8 208002.8 364609.8 360908.9 NA## FT005 NA NA NA 107348.5 223951.8 NA NA## FT006 286221.4 NA 164009.0 149097.6 255697.7 311296.8 366441.5## wt22.CDF## FT001 NA## FT002 1354004.93## FT003 185399.47## FT004 221937.53## FT005 84772.92## FT006 271128.02
可以使用数据imputation来解决(使用 fillChromPeaks 函数),该方法将feature所在的m/z-RT区域测量到的信号集成到报告了NA的原始样本文件中,从而填补缺失的峰数据。
算法
-
ChromPeakAreaParam:根据已识别特征色谱峰(chromatographic peaks of the feature)的m / z和rt范围定义区域 -
FillChromPeaksParam: uses columns"mzmin","mzmax","rtmin"and"rtmax"in thefeatureDefinitionsdata frame.
峰补齐代码:
xdata <- fillChromPeaks(xdata, param = ChromPeakAreaParam())
head(featureValues(xdata))## ko15.CDF ko16.CDF ko21.CDF ko22.CDF wt15.CDF wt16.CDF wt21.CDF## FT001 159738.1 506848.88 113441.08 169955.6 216096.6 145509.7 230477.9## FT002 1924712.0 1757150.96 1383416.72 1180288.2 2129885.1 1634342.0 1623589.2## FT003 213659.3 289500.67 162897.19 178285.7 253825.6 241844.4 240606.0## FT004 349011.5 451863.66 343897.76 208002.8 364609.8 360908.9 223322.5## FT005 135978.5 25524.79 71530.84 107348.5 223951.8 134398.9 190203.8## FT006 286221.4 289908.23 164008.97 149097.6 255697.7 311296.8 366441.5## wt22.CDF## FT001 140551.30## FT002 1354004.93## FT003 185399.47## FT004 221937.53## FT005 84772.92## FT006 271128.02
吐槽
-
IPO调优的时间特别特别特别特别特别特别特别的长。
-
IPO在使用除demo数据外的格式,报错不说,还直接把R退出来了(亲测了一个只包含1级质谱数据的mzML/mzXML(两个格式都试了)的数据,含泪放弃)。
参考资料
-
Automated Optimization of XCMS Data Processing parameters
-
Metabolomics data pre-processing using xcms
-
LCMS data preprocessing and analysis with xcms
以上是关于非靶代谢组学数据预处理XCMS+IPO的主要内容,如果未能解决你的问题,请参考以下文章