转录组实战01: 从数据下载到定量fastp+STAR
Posted 生信探索
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了转录组实战01: 从数据下载到定量fastp+STAR相关的知识,希望对你有一定的参考价值。
生信交流与合作请关注公众号@生信探索
01.建立工作目录
cd ~
mkdir -p Project/Human_16_Asthma_Bulk
cd Project/Human_16_Asthma_Bulk
# 建立数据存放目录 data
mkdir -p data/rawdata data/cleandata/fastp
# 建立STAR目录
mkdir STAR
02. 环境准备
micromamba create -n RNA
micromamba activate RNA
micromamba install -y -c hcc aspera-cli
micromamba install -y -c bioconda fastqc multiqc fastp samtools star
03.准备文件
https://www.gencodegenes.org/human/
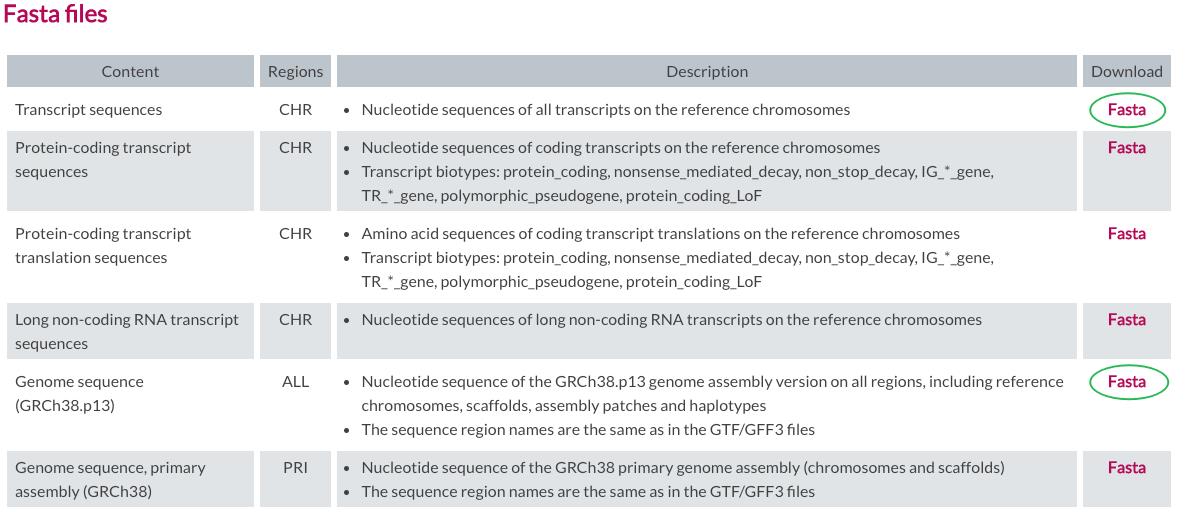

mkdir -p ~/DataHub/Genomics/GENCODE/release_42
cd ~/DataHub/Genomics/GENCODE/release_42
# 下载参考基因组及注释文件
#>>>downGENCODE.sh
wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_42/gencode.v42.annotation.gtf.gz
wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_42/GRCh38.p13.genome.fa.gz
mv gencode.v42.annotation.gtf.gz HS.gencode.v42.annotation.gtf.gz
mv GRCh38.p13.genome.fa.gz HS.GRCh38.p13.genome.fa.gz
gzip -kd HS.gencode.v42.annotation.gtf.gz
gzip -kd HS.GRCh38.p13.genome.fa.gz
#<<<downGENCODE.sh
nohup zsh downGENCODE.sh &> downGENCODE.sh.log &
04.建立索引
-
构建star的索引
mkdir -p ~/DataHub/Genomics/star_index/human
cd ~/DataHub/Genomics/star_index/human
n_jobs=12
#>>>star_index.sh>>>
STAR \\
--runMode genomeGenerate \\
--genomeDir ~/DataHub/Genomics/star_index/human \\
--genomeFastaFiles ~/DataHub/Genomics/GENCODE/release_42/HS.GRCh38.p13.genome.fa \\
--sjdbOverhang 100 \\
--sjdbGTFfile ~/DataHub/Genomics/GENCODE/release_42/HS.gencode.v42.annotation.gtf \\
--runThreadN $n_jobs
#<<<star_index.sh<<<
nohup sh star_index.sh &> star_index.sh.log &
05.下载原始数据
https://www.ebi.ac.uk/ena/browser/view/PRJNA229998,下载TSV文件
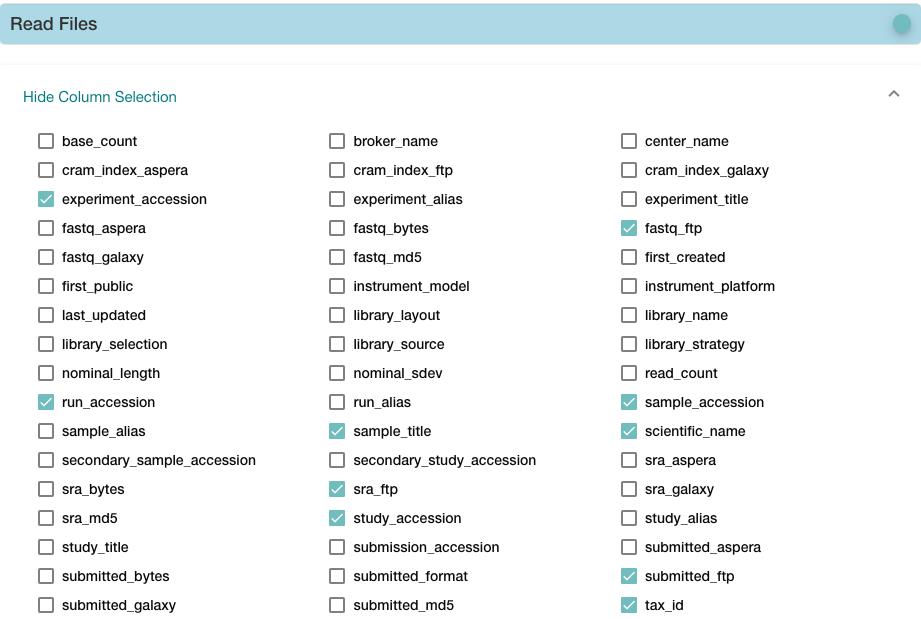
cd ~/Project/Human_16_Asthma_Bulk/data/rawdata
cut -f 9 filereport_read_run_PRJNA229998_tsv.txt | tr ';' '\\n' |grep '_[12].fastq.gz' > fq.txt
#>>>downloadFQ.sh
cat fq.txt |while read i
do
ascp -QT -l 300m -P33001 \\
-i ~/micromamba/envs/RNA/etc/asperaweb_id_dsa.openssh \\
era-fasp@$i \\
.
done
#<<<downloadFQ.sh
nohup sh downloadFQ.sh &> downloadFQ.log &
#制作了check.md5文件
cut -f 7 filereport_read_run_PRJNA229998_tsv.txt | sed '1d' | awk -F ';' 'print $(NF-1)"\\n"$NF' > md5.txt
cut -f 7 -d '/' fq.txt | paste md5.txt - > check.md5
rm md5.txt
#检查md5值是否一样,文件是否下载完整
d5sum -c check.md5
05.质控
质量评估
#fastqc
nohup fastqc -t 6 -o . SRR*.fastq.gz > fastqc.log &
#multiqc
multiqc *.zip
fastp数据过滤
#>>>fastp.sh>>>
rawdata_dir=~/Project/Human_16_Asthma_Bulk/data/rawdata
cleandata_dir=~/Project/Human_16_Asthma_Bulk/data/cleandata/fastp
n_jobs=8
cut -f 4 $rawdata_dir/filereport_read_run_PRJNA229998_tsv.txt | sed '1d' > $rawdata_dir/run_accession.txt
cat $rawdata_dir/run_accession.txt | while read i
do
fastp \\
--in1 $rawdata/$i_1.fastq.gz \\
--in2 $rawdata/$i_2.fastq.gz \\
--out1 $cleandata/$i_1.fastp.fq.gz \\
--out2 $cleandata/$i_2.fastp.fq.gz \\
--json $cleandata/$i.fastp.json \\
--html $cleandata/$i.fastp.html \\
--report_title $cleandata/$i \\
--thread $n_jobs
done
#<<<fastp.sh<<<
cd ~/Project/Human_16_Asthma_Bulk/data/cleandata/fastp
nohup sh fastp.sh &> fastp.sh.log &
06.STAR数据比对和定量
-
统计对比结果
#>>>star.sh>>>
rawdata_dir=~/Project/Human_16_Asthma_Bulk/data/rawdata
index_dir=~/DataHub/Genomics/star_index/human
input_dir=~/Project/Human_16_Asthma_Bulk/data/cleandata/fastp
out_dir=~/Project/Human_16_Asthma_Bulk/STAR
n_jobs=8
cat $rawdata_dir/run_accession.txt | while read i
do
STAR \\
--readFilesIn $input_dir/$i_1.fastp.fq.gz $input_dir/$i_2.fastp.fq.gz \\
--outSAMattrRGline ID:sample SM:sample PL:ILLUMINA \\
--genomeDir $index_dir \\
--readFilesCommand zcat \\
--runThreadN $n_jobs \\
--twopassMode Basic \\
--outFilterMultimapNmax 20 \\
--alignSJoverhangMin 8 \\
--alignSJDBoverhangMin 1 \\
--outFilterMismatchNmax 999 \\
--outFilterMismatchNoverLmax 0.1 \\
--alignIntronMin 20 \\
--alignIntronMax 1000000 \\
--alignMatesGapMax 1000000 \\
--outFilterType BySJout \\
--outFilterScoreMinOverLread 0.33 \\
--outFilterMatchNminOverLread 0.33 \\
--limitSjdbInsertNsj 1200000 \\
--outFileNamePrefix $out_dir/$i \\
--outSAMstrandField intronMotif \\
--outFilterIntronMotifs None \\
--alignSoftClipAtReferenceEnds Yes \\
--quantMode TranscriptomeSAM GeneCounts \\
--outSAMtype BAM SortedByCoordinate \\
--outSAMunmapped Within \\
--genomeLoad NoSharedMemory \\
--chimSegmentMin 15 \\
--chimJunctionOverhangMin 15 \\
--chimOutType Junctions SeparateSAMold WithinBAM SoftClip \\
--chimOutJunctionFormat 1 \\
--chimMainSegmentMultNmax 1 \\
--outSAMattributes NH HI AS nM NM ch
done
#<<<star.sh<<<
nohup sh star.sh &> star.sh.log &
整理count数据,想不出怎么用linux合并数据框比较优雅,所以用Python吧还是
from pathlib import Path
import pandas as pd
import datatable as dt
dir="STAR"
count_list = []
tpm_list = []
paths = Path(dir).glob("*ReadsPerGene.out.tab")
for x,y in enumerate(list(paths)):
if x < 1:
Geneid = pd.read_csv(y,usecols=[0],sep='\\t',skiprows=4,header=None)
Geneid.rename(columns=0:'Geneid',inplace=True)
_count_df = pd.read_csv(y,usecols=[1],sep='\\t',skiprows=4,header=None)
count_list.append(_count_df.rename(columns=1:y.name.split('Reads')[0]))
count_df = pd.concat(count_list,axis=1)
count_df.insert(0,column='Geneid',value=Geneid)
dt.Frame(count_df).to_csv('star_count.csv.gz')
-
对比质量
#>>>star_qc.sh>>>
cd ~/Human_16_Asthma_Bulk/STAR
n_jobs=12
ls *sortedByCoord.out.bam | while read i
do
samtools flagstat -@ $n_jobs $i > $i/bam/flagstat
done
multiqc -o ./ *.flagstat
#<<<star_qc.sh<<<
nohup zsh star_qc.sh &> star_qc.sh.log &
Reference
https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
https://github.com/OpenGene/fastp
https://www.jianshu.com/p/2fa4f5b870f5
https://www.jianshu.com/p/6da36135e2d1
https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
https://zhuanlan.zhihu.com/p/360427232
https://www.jianshu.com/p/2fa4f5b870f5
https://zhuanlan.zhihu.com/p/345896914
https://mp.weixin.qq.com/s/OJ1MpHhW08BcZSjMvFs3eg
https://mp.weixin.qq.com/s/OJ1MpHhW08BcZSjMvFs3eg
环境
List of packages in environment: "~/micromamba/envs/RNA"
Name Version Build Channel
──────────────────────────────────────────────────────────────────────────────────────────
_libgcc_mutex 0.1 main anaconda/pkgs/main
_openmp_mutex 5.1 1_gnu anaconda/pkgs/main
aspera-cli 3.9.6 h5e1937b_0 hcc
backports 1.1 pyhd3eb1b0_0 anaconda/pkgs/main
backports.functools_lru_cache 1.6.4 pyhd3eb1b0_0 anaconda/pkgs/main
backports_abc 0.5 py_1 anaconda/pkgs/main
blas 1.0 mkl anaconda/pkgs/main
bz2file 0.98 py27_1 anaconda/pkgs/main
bzip2 1.0.8 h7b6447c_0 anaconda/pkgs/main
c-ares 1.18.1 h7f8727e_0 anaconda/pkgs/main
ca-certificates 2022.10.11 h06a4308_0 anaconda/pkgs/main
certifi 2020.6.20 pyhd3eb1b0_3 anaconda/pkgs/main
click 7.1.2 pyhd3eb1b0_0 anaconda/pkgs/main
curl 7.87.0 h5eee18b_0 anaconda/pkgs/main
cutadapt 1.18 py27h14c3975_1 anaconda/cloud/bioconda
cycler 0.10.0 py27_0 anaconda/pkgs/main
dbus 1.13.18 hb2f20db_0 anaconda/pkgs/main
expat 2.4.9 h6a678d5_0 anaconda/pkgs/main
fastp 0.22.0 h2e03b76_0 anaconda/cloud/bioconda
fastqc 0.11.9 hdfd78af_1 anaconda/cloud/bioconda
font-ttf-dejavu-sans-mono 2.37 hd3eb1b0_0 anaconda/pkgs/main
fontconfig 2.14.1 h52c9d5c_1 anaconda/pkgs/main
freetype 2.12.1 h4a9f257_0 anaconda/pkgs/main
functools32 3.2.3.2 py27_1 anaconda/pkgs/main
futures 3.3.0 py27_0 anaconda/pkgs/main
gdbm 1.18 hd4cb3f1_4 anaconda/pkgs/main
gettext 0.21.0 hf68c758_0 anaconda/pkgs/main
git 2.34.1 pl5262hc120c5b_0 anaconda/pkgs/main
glib 2.69.1 h4ff587b_1 anaconda/pkgs/main
gst-plugins-base 1.14.0 h8213a91_2 anaconda/pkgs/main
gstreamer 1.14.0 h28cd5cc_2 anaconda/pkgs/main
hisat2 2.1.0 py27hc9558a2_4 anaconda/cloud/bioconda
htslib 1.13 h9093b5e_0 anaconda/cloud/bioconda
icu 58.2 he6710b0_3 anaconda/pkgs/main
intel-openmp 2022.1.0 h9e868ea_3769 anaconda/pkgs/main
jinja2 2.11.3 pyhd3eb1b0_0 anaconda/pkgs/main
jpeg 9e h7f8727e_0 anaconda/pkgs/main
kiwisolver 1.1.0 py27he6710b0_0 anaconda/pkgs/main
krb5 1.19.2 hac12032_0 anaconda/pkgs/main
libcurl 7.87.0 h91b91d3_0 anaconda/pkgs/main
libdeflate 1.7 h27cfd23_5 anaconda/pkgs/main
libedit 3.1.20210714 h7f8727e_0 anaconda/pkgs/main
libev 4.33 h7f8727e_1 anaconda/pkgs/main
libffi 3.3 he6710b0_2 anaconda/pkgs/main
libgcc-ng 11.2.0 h1234567_1 anaconda/pkgs/main
libgfortran-ng 7.5.0 ha8ba4b0_17 anaconda/pkgs/main
libgfortran4 7.5.0 ha8ba4b0_17 anaconda/pkgs/main
libgomp 11.2.0 h1234567_1 anaconda/pkgs/main
libnghttp2 1.46.0 hce63b2e_0 anaconda/pkgs/main
libpng 1.6.37 hbc83047_0 anaconda/pkgs/main
libssh2 1.10.0 h8f2d780_0 anaconda/pkgs/main
libstdcxx-ng 11.2.0 h1234567_1 anaconda/pkgs/main
libuuid 1.41.5 h5eee18b_0 anaconda/pkgs/main
libxcb 1.15 h7f8727e_0 anaconda/pkgs/main
libxml2 2.9.14 h74e7548_0 anaconda/pkgs/main
markupsafe 1.1.1 py27h7b6447c_0 anaconda/pkgs/main
matplotlib 2.2.3 py27hb69df0a_0 anaconda/pkgs/main
mkl 2020.2 256 anaconda/pkgs/main
mkl-service 2.3.0 py27he904b0f_0 anaconda/pkgs/main
mkl_fft 1.0.15 py27ha843d7b_0 anaconda/pkgs/main
mkl_random 1.1.0 py27hd6b4f25_0 anaconda/pkgs/main
multiqc 0.9.1a0 py27_4 anaconda/cloud/bioconda
ncurses 6.2 he6710b0_1 anaconda/pkgs/main
numpy 1.16.6 py27hbc911f0_0 anaconda/pkgs/main
numpy-base 1.16.6 py27hde5b4d6_0 anaconda/pkgs/main
openjdk 11.0.13 h87a67e3_0 anaconda/pkgs/main
openssl 1.1.1s h7f8727e_0 anaconda/pkgs/main
pcre 8.45 h295c915_0 anaconda/pkgs/main
pcre2 10.37 he7ceb23_1 anaconda/pkgs/main
perl 5.34.0 h5eee18b_2 anaconda/pkgs/main
pigz 2.6 h27cfd23_0 anaconda/pkgs/main
pip 19.3.1 py27_0 anaconda/pkgs/main
pyparsing 2.4.7 pyhd3eb1b0_0 anaconda/pkgs/main
pyqt 5.9.2 py27h05f1152_2 anaconda/pkgs/main
python 2.7.18 ha1903f6_2 anaconda/pkgs/main
python-dateutil 2.8.2 pyhd3eb1b0_0 anaconda/pkgs/main
pytz 2021.3 pyhd3eb1b0_0 anaconda/pkgs/main
pyyaml 5.2 py27h7b6447c_0 anaconda/pkgs/main
qt 5.9.7 h5867ecd_1 anaconda/pkgs/main
readline 8.1 h27cfd23_0 anaconda/pkgs/main
samtools 1.13 h8c37831_0 anaconda/cloud/bioconda
setuptools 44.0.0 py27_0 anaconda/pkgs/main
simplejson 3.8.1 py27_0 anaconda/cloud/bioconda
singledispatch 3.7.0 pyhd3eb1b0_1001 anaconda/pkgs/main
sip 4.19.8 py27hf484d3e_0 anaconda/pkgs/main
six 1.16.0 pyhd3eb1b0_1 anaconda/pkgs/main
sqlite 3.40.1 h5082296_0 anaconda/pkgs/main
star 2.7.10b h9ee0642_0 anaconda/cloud/bioconda
subprocess32 3.5.4 py27h7b6447c_0 anaconda/pkgs/main
subread 2.0.1 h5bf99c6_1 anaconda/cloud/bioconda
tbb 2020.3 hfd86e86_0 anaconda/pkgs/main
tk 8.6.12 h1ccaba5_0 anaconda/pkgs/main
tornado 5.1.1 py27h7b6447c_0 anaconda/pkgs/main
trim-galore 0.6.7 hdfd78af_0 anaconda/cloud/bioconda
wheel 0.37.1 pyhd3eb1b0_0 anaconda/pkgs/main
xopen 0.7.3 py_0 anaconda/cloud/bioconda
xz 5.2.10 h5eee18b_1 anaconda/pkgs/main
yaml 0.1.7 had09818_2 anaconda/pkgs/main
zlib 1.2.13 h5eee18b_0 anaconda/pkgs/main
本文由 mdnice 多平台发布
以上是关于转录组实战01: 从数据下载到定量fastp+STAR的主要内容,如果未能解决你的问题,请参考以下文章
lncRNA数据处理(get fasta>>>novel lncRNA)))