测序原理笔记 RNA-seq 和WES--day 5
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了测序原理笔记 RNA-seq 和WES--day 5相关的知识,希望对你有一定的参考价值。
参考技术A 1,RNA-seq 测试原理测序步骤,先去除保守的rRNA ,→polyA 尾的特性,用磁珠吸附→Mg镁离子溶液打断mRNA→随机引物变成双链合成第一条cDNA(由RNA变成单链DNA)→再合成第二条DNA(双链DNA)→用相应的酶切加上A,加上Y 行接头(adaptor)
主要应用:差异表达mRNA,可变剪切,融合基因,SNP,
RNA 降解评价,RNA的RIN(RNA integrity number )> 8 分,比较好;
2,WES 测序步骤
总体的思路是用 外显子探针捕获库 去杂交外显子,然后进行pcr扩增,然后进行测序;
steps: 基因组被打断成片段建成 DNA文库→用结合了 生物素探针的捕获试剂盒进行捕获→用链霉素生物素结合磁珠捕获生物素 →洗脱磁珠上的序列→PCR扩增→测序
主要的应用:发现SNP ,插入缺失突变也就是Indel 信息;不适用:基因结构变异(structure variation,SV),对拷贝数变异(copy number variation (CNV))不敏感,如果是大片段的拷贝数变异,比如5M 以上,因为大片段会出现多个外显子,(全基因组测序,发现SV 和CNV),不能发现基因表达的差异;
3,名词解释
biotin ,生物素;streptavidin 链霉亲和素
比较: WES,RNA-seq,这几个测序的差异
单端测序 和双端测序
Illumina测序原理
由于现在手头所分析的RNA-seq要用到的数据全部是来自与Illumia测序,所以决定先看看Illumina的测序原理。
决定先看youtube上的陈巍学基因。
https://www.youtube.com/watch?v=VvS8NEJGxnM
其中的一些注意点:
1)adapter是接头,分为pcr扩增接头和测序接头,分别用于文库构建和测序的引物。
index是索引序列,为区分不同样本而构建。高通量测序一次一块板要加好多样品,如果你的样品少那单独测你的测序公司就亏了,因此把你的和别人的样品放到一起,分别加个index用以区分,测序结束后就可以根据index来分离单个样本了
2)双端测序,应该是先从第一链的接头那里开始测,测完后,从第二链的接头那里开始测,并非是在一条链上两端测序
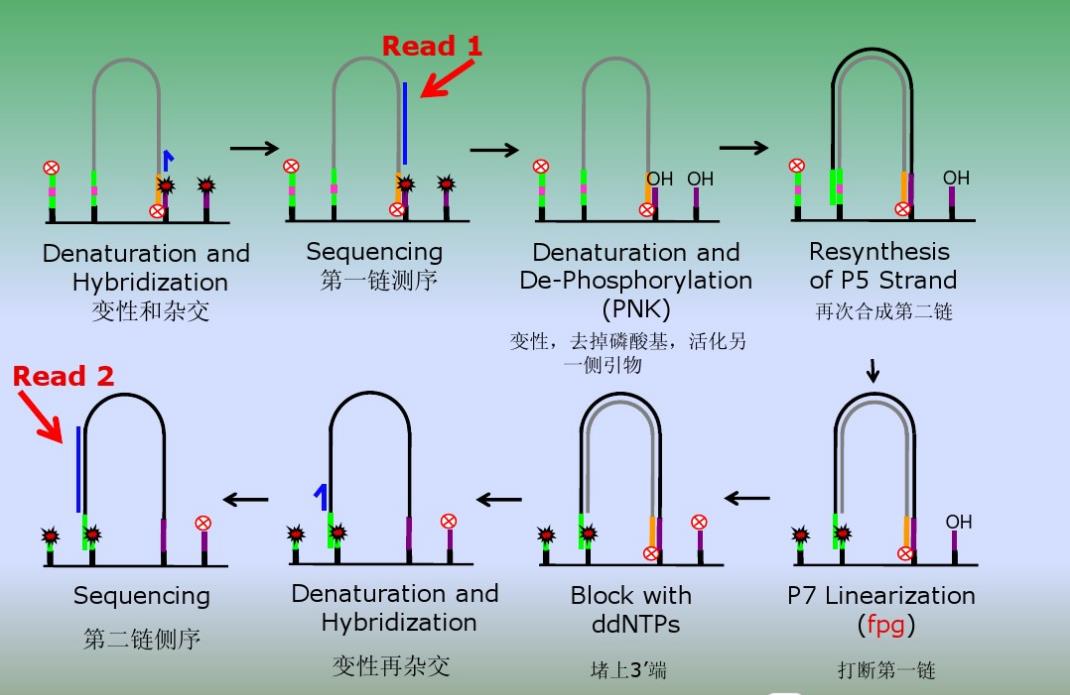
3)Pair-end reads,就是双端测序,简称PE,也就是一段DNA,从正向读一次,再从反向读一次。与双端测序相对的,是单端测序,也叫Sing read,简称SR,在中国大陆,也有许多人将之称为SE,single-end。
4)1、测序深度:测序得到的总碱基数与待测基因组大小的比值。假设一个基因组大小为7M,测序总碱基数为70M,则测序深度为10×。
2、覆盖度:测序获得的序列占整个基因组的比例。由于基因组中高GC含量,重复序列等复杂结构的存在,测序最终拼接组装的序列往往无法覆盖所有的区域,这些区域就叫做Gap。
二者的关系:测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降。当测序深度在10~15X以上时,基因组覆盖度和测序错误率控制均得以保证。
5)RPKM(reads per kilobase per million reads)是每百万读段中来自于某基因每千碱基长度的读段数。其公式为:

其中,total exon reads指映射到某个基因上的reads数,mapped reads指map到所有基因的总的reads数。
RPKM不仅对测序深度作了归一化,而且对基因长度也作了归一化,使得不同长度的基因在不同测序深度下得到的基因表达水平估计值具有了可比性,是目前最常用的基因表达估计方法。
之所以分母还要加上这个基因的外显子长度。因为基因是被打断从而测序的,如果一个基因的外显子越长,则其所产生的RNA就越长,被打出来的小片段就越多,比如A基因,1kb,则其可能会被打成5个200bp,而B基因,2kb,则10个200bp,所以B基因在测序中被测到的概率会比A高一倍。除以外显子长度,是为了修正mRNA长度所引起的mRNA的read数的偏差。
以上是关于测序原理笔记 RNA-seq 和WES--day 5的主要内容,如果未能解决你的问题,请参考以下文章