PacBio的异构体测序(Iso-Seq)
Posted ryannbio
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了PacBio的异构体测序(Iso-Seq)相关的知识,希望对你有一定的参考价值。
在真核生物中,大多数的基因可以编码多个蛋白质,这是因为基因经过可变剪接,可产生多个转录异构体,从而大大增加基因组的蛋白编码潜力。来自同一个基因的可变剪接异构体可能有着明显不同、甚至拮抗的作用。为了研究基因表达,研究人员利用新一代测序方法研究了生物体各个基因的片段,这种方法通常称为RNA测序(RNA-seq)。短读长RNA-seq的原理是将转录本异构体打断成较小的片段,然后利用生物信息学工具将其重新组装。由于组装错误的存在,RNA-seq很可能无法获得完整的转录本,因而难以准确表征异构体的多样性。
PacBio的异构体测序(Iso-Seq)采用长读取序列来测序长达10 kb的转录本异构体。无论是广泛研究还是靶向分析,这种转录本多样性的分析都揭示了可变转录的频率和类型等关键信息,改善了基因组注释和基因发掘。
Iso-seq方法
Iso-Seq无需打断RNA分子,直接对反转录的全长cDNA测序,可提供从5’端到3′ polyA尾巴、跨越整个转录本异构体的序列。Iso-Seq方法可提供选择性剪接外显子和转录起始位点的准确信息。对于长达10 kb的转录本,它还可提供聚腺苷酸化位点的信息,能够覆盖靶基因或整个转录组的全长异构体。
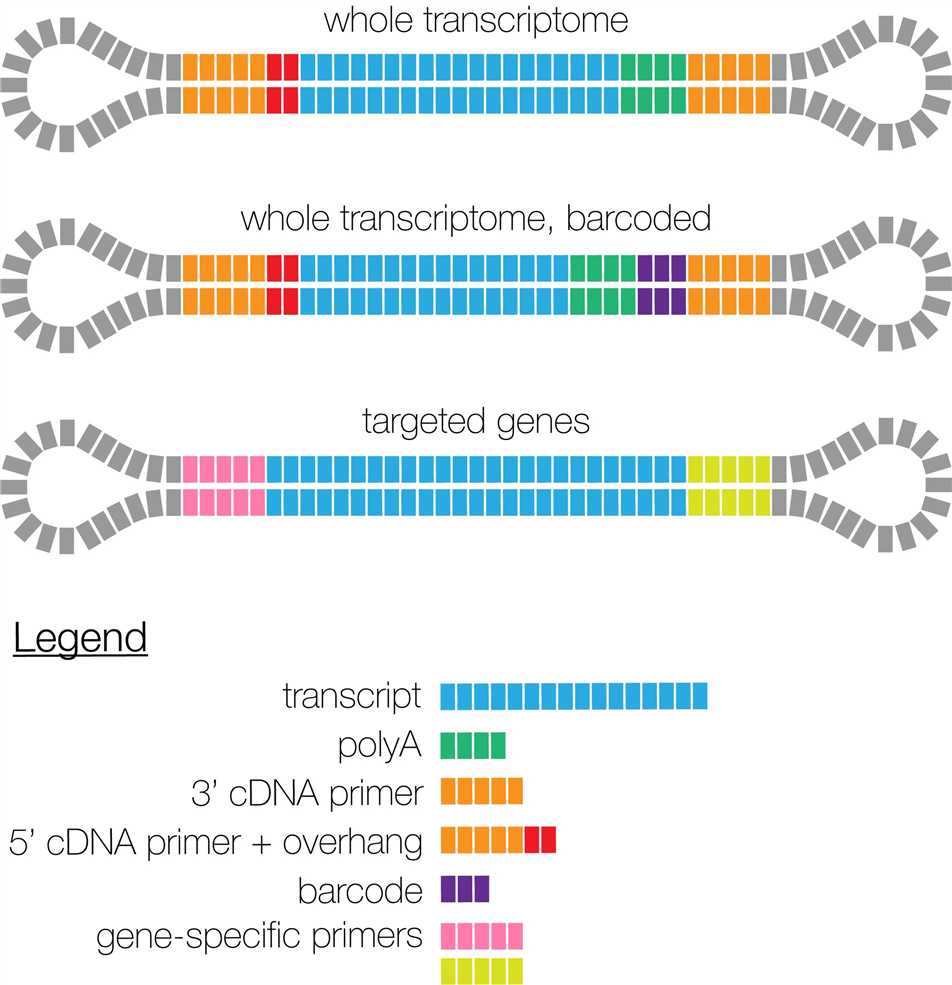
Iso-Seq的建库方案有如下三类:
- 整个库都是一个样品的全长转录组,不需要加barcode区分样品
- 不同样品的全长转录组,加上不同barcode ,可以放在一起进行建库测序
- 一些靶向获得的部分基因也可以进行全长转录组的测序
Iso-seq分析
Pacbio sequel下机是bam格式的reads文件,它和reads比对到参考基因组上生成的bam文件,内容有差异,但格式一致。格式说明可参考(https://www.plob.org/article/11099.html)。
RS_IsoSeq.1
该应用程序适用于分析SMRT测序技术生成的数据,能够对转录本和剪接变体进行功能鉴定。
Iso-Seq分析运行可选择从头开始(de novo)或基于参考序列的模式运行。
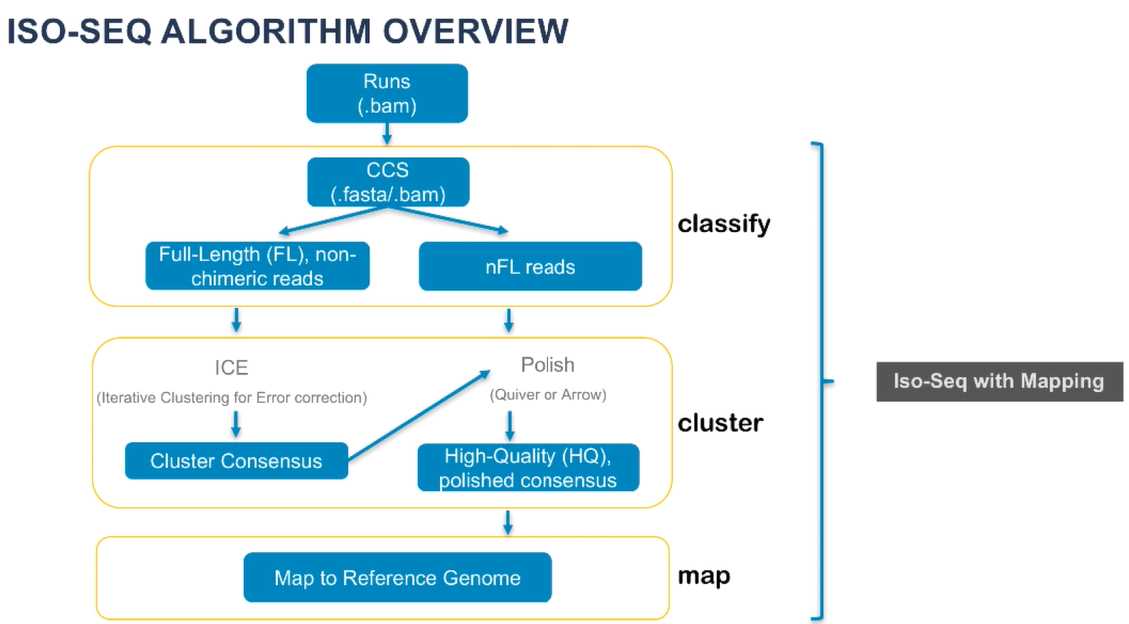
它包括三个主要步骤:
- 分类:从PacBio系统(或SMRT Cell)运行中提取插入片段的序列;去除cDNA引物和poly-A;然后将插入片段的读取序列分成嵌合或非嵌合、全长或非全长的序列。
- 聚类:利用迭代聚类和错误纠正(ICE)算法,根据分类的读取序列预测新发的转录本一致性异构体。
- 映射:利用GMAP,将分类的读取序列和预测的一致性异构体与用户指定的参考序列进行比对。
以上是关于PacBio的异构体测序(Iso-Seq)的主要内容,如果未能解决你的问题,请参考以下文章