RNA测序究竟有多可靠
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了RNA测序究竟有多可靠相关的知识,希望对你有一定的参考价值。
(转载)RNA测序在生物科学和医学研究中非常流行,而且正在逐渐走向临床应用。那么RNA测序到底可不可靠呢?由美国FDA牵头的测序质量控制(SEQC)项目对RNA测序的准确性、可重现性和信息含量进行了综合性评估,并将初步调查结果发表在Nature Biotechnology杂志上。
RNA测序可以检测人类和其他生物的基因表达情况。最近这一方法在生物科学和医学研究中非常流行,而且正在逐渐走向临床应用。与之前的方法相比,RNA测序的优势是便于研究选择性剪切形成的基因异构体或转录本。
那么RNA测序到底可不可靠呢?日前,由美国FDA牵头的测序质量控制(SEQC)项目对RNA测序的准确性、可重现性和信息含量进行了综合性评估,并将初步调查结果发表在近日的Nature Biotechnology杂志上。石乐明教授是这篇文章的通讯作者之一。
研究团队使用RNA参照样本,在全球多个实验室的Illumina HiSeq、Life Technologies SOLiD、Roche 454平台上进行了检测。研究人员主要是评估RNA测序在接头区域和差异性表达谱中的表现,并将其与芯片和定量PCR(qPCR)进行比较。
研究人员发现,所有测序深度都会出现未注释的外显子-外显子连接区域,其中80%以上都得到了qPCR的验证。用RNA测序检测相对表达可以得到准确且可重复的结果,但RNA测序和芯片都不能提供精确的绝对测量,而且研究用到的平台都存在基因特异性的偏好,包括qPCR。
数据分析的算法也会对RNA测序产生很大影响,不同算法生成的转录本数据差异很大。研究显示,赫尔辛基大学和曼彻斯特大学开发的BitSeq能生成最可靠的结果,这一方法以概率建模为基础。 参考技术A 你要看是什么RNA~普通转录组mRNA技术还是很成熟的~基本没什么问题~其他特殊一些的比如microRNA circleRNA lncRNA等可靠性会低一些~
第三章 RNA测序
RNA测序(RNA Sequencing,简称RNA-Seq,也被称为全转录物组鸟枪法测序Whole Transcriptome Shotgun Sequencing,简称WTSS),是基于二代测序技术研究转录组学的方法,可以快速获取给定时刻的一个基因组中RNA的种类和数量。

-
RNA-Seq有助于查看基因的不同转录本、转录后修饰、基因融合、突变/SNP和基因表达随时间的变化,或在不同组中基因表达的差异。
-
RNA-Seq除了可以查看mRNA转录本,还可以查看总RNA、小RNA,例如miRNA、tRNA和核糖体RNA。
-
RNA-Seq可用于确定外显子/内含子边界,并验证或修正已注释的5‘和3‘基因边界。
-
RNA-Seq最新的研究包括单细胞测序和固定组织的原位测序。
文库制备
RNA的cDNA文库制备通常包括如下几个步骤:RNA提取和分离、RNA类型选择和消化、cDNA合成。但不同的平台可能会有所不同。

分析
转录本组装
有两种策略将测序数据用于转录本组装:
- 重头组装:这种方法不需要参考基因组来重组转录组,并且通常用于基因组未知、不完整或者差别很大时使用。使用测序read从头组装时面临的挑战包括:1)遇到重叠群时确定哪些片段应该连接在一起成为连续序列; 2)测序错误或人工错误的不稳定性;3)计算效率。从头组装的主要算法从重叠图转换为de Bruijn图,使用de Bruijn图的汇编程序有Velvet、Trinity、Oases和Bridger。评估从头组装质量指标包括中位重叠群长度、重叠群数量和N50。
- 基于参考基因组组装:这种方法依赖于序列比对算法,比对的read覆盖了参考基因组,产生非连续部分,这些非连续的read是对成熟mRNA测序的结果(见图)。通常,比对算法有两个步骤:1)用read的短序列进行比对;2)用动态编程找到最佳比对,有时结合已知的注释。基于基因组比对的软件工具包括Bowtie、TopHat(基于Bowtie比对结果对齐剪切点)、Subread、STAR、Sailfish、Kallisto和GMAP。 评估基于参考基因组组装质量指标主要是以下两点:1)从头组装指标(例如,N50);2)与已知转录本、剪切点、基因组和蛋白序列等进行比较。
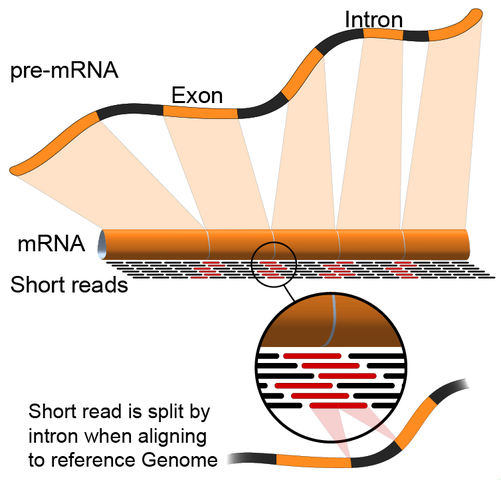
关于组装质量,目前的情况是:1)装备质量根据使用的标准而变化;2)在一个物种中得分良好的软件在其他物种中不一定表现良好;3)组合使用不同软件可能是最可靠的。
基因表达
量化表达通常用于研究响应外部刺激的细胞变化、健康和患病状态之间的差异以及其它研究问题。基因表达通常用来反映蛋白质丰度,但不适用于诸如RNA干扰和无义介导的转录衰变事件。
通过统计转录组装配步骤中映射到每个基因座的read数量来量化表达,使用重叠群或注释的转录本来定量外显子或基因的表达,这种统计RNA-Seq read数的方法已经通过较老的技术(表达微列阵和qPCR)进行了有效地验证。量化计数工具有HTSeq、FeatureCounts、Rcount、maxcounts、FIXSEQ和Cuffquant,这些工具都是将统计read数转换为适用于假设检验、回归和其它分析的指标。此状换参数是:
文库大小:虽然在进行多个RNA-Seq实验时预先确定了测序深度,但实验之间仍会有很大差异。因此,通常将read统计数转换为每百万比对read的read数、片段数或个数(FPM、RPM或CPM)来调整在单个实验中生成的read总数(文库大小)。
基因长度:如果转录本表达相同,则较长的基因将比较短的基因具有更多的片段、read或个数。通过将FPM除以基因长度来调整,得到每千个碱基的转录每百万比对read的片段(FPKM)。当查看样本间的基因组时,通过将每个FPKM除以样本中FPKM的总和,转换为百万分之一的转录本(TPM)。
样本总RNA:因为每个样本中提取相同量的RNA,样本总RNA多的将具有更少种类基因RNA,导致下游分析中的假阳性。
每个基因的表达方差:建模以考虑抽样误差(对具有低read数的基因很重要),可以将方差估计为正常、泊松或负二项分布。
转录本的差异表达和绝对定量
RNA-Seq通常用于比较不同条件之间的基因表达,例如药物治疗与未治疗,并找出每种情况下哪些基因上调或下调。理论上,RNA-Seq可以统计每种情况下细胞中的所有转录本的个数,通过测序read统计工具统计每个基因的read数,并在样本间进行比较来鉴定不同表达的基因。有许多软件包可用于此类分析,常用的工具是来自Bioconductor软件包DESeq和edgeR,这两个个工具都使用基于负二项分布的模型。
一般的RNA-Seq分析不能进行绝对定量,因为它仅提供相对于所有转录本的RNA水平,如果细胞中RNA总量随不同条件发生变化,则相对标准化将错误地表示个体转录本的变化。通过添加外标(已知浓度的RNA样品)进行RNA-Seq,可以对mRNA进行绝对定量。
基因共表达网络分析
基因共表达网络分析是根据基因表达量的动态变化,计算基因间的共表达关系,来建立基因转录调控模型,得到基因间的表达调控关系及调控方向,从而寻找一个或多个物种在不同发育阶段,或者不同组织在不同条件或处理下的全部基因表达调控网络模型以及关键基因。
发现单核苷酸突变(SNP)
RNA-Seq仅限于发现外显子区域的序列变异,不能检测到内含子区域序列变异。虽然外显子与内含子变异之间存在某种相关性,但只有全基因组测序才能捕获所有来源的SNP。
绝对确定个体突变的方法是将转录本序列与种系DNA进行比对。这样能够区分纯合基因与其中一个等位基因的倾斜表达,并且还可以提供关于转录组实验中未表达的基因的信息。一个基于R语言名为CummeRbund包可用于生成视觉化的表达图表。
RNA编辑(转录后改变)
比较个体的基因组和转录组序列也可以帮助检测转录后编辑,如果基因是纯合的,但是基因具有不同的转录本,则确定是转录后修饰。
融合基因检测
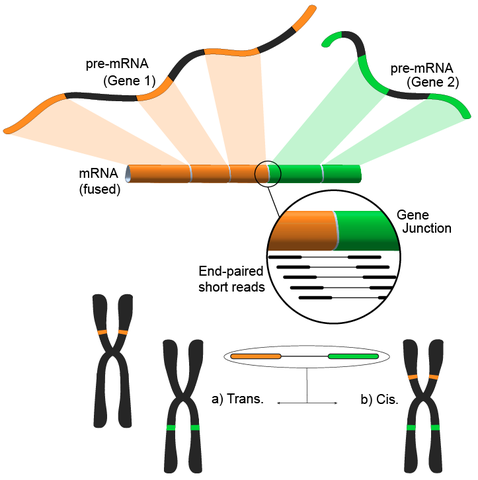
由于基因组中的不同结构修饰,融合基因因其与癌症的关系而受到关注。RNA-Seq因无差别分析整个转录组的能力使其成为癌症研究中发现这些常见事件的有力工具。
该方法遵循将短转录组read比对到参考基因组的过程,大多数短read将比对到一个完整的外显子内,仍有一大部分比对到已知的外显子——外显子连接,然后进一步分析剩余未必对的read是否匹配到外显子——来自不同基因的外显子连接,这可能是融合事件的有力证据,然而,由于read的长度,事实上是比较粗糙的方法。另一种方法是使用双端read,当潜在大量双端read将每个末端比对到不同的外显子时,可以更好地验证这些事件(见图)。
应用
-
转录本结构研究(基因边界鉴定、可变剪切研究等)
-
转录本变异研究(如基因融合、编码区SNP研究)
-
非编码区域功能研究(Non-coding RNA、microRNA前体研究等)
-
基因表达水平研究以及全新转录本发现
参考资料
以上是关于RNA测序究竟有多可靠的主要内容,如果未能解决你的问题,请参考以下文章