无生物学重复RNA-seq分析 CORNAS: coverage-dependent RNA-Seq analysis of gene expression data without biologi
Posted wangprince2017
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了无生物学重复RNA-seq分析 CORNAS: coverage-dependent RNA-Seq analysis of gene expression data without biologi相关的知识,希望对你有一定的参考价值。
无生物学重复RNA-seq分析
CORNAS: coverage-dependent RNA-Seq analysis of gene expression data without biological replicates
BMC Bioinformatics 的一篇文章中提出了一种新的差异基因分析方法。
这篇文章提出了CORNAS(COverage-dependent RNA-Seq) 方法,利用贝叶斯方法来推断真实基因表达数的 后验分布。
其创新型之一该方法包括了由RNA样品浓度决定的覆盖度参数,之二是真实基因表达量后验分布的比较为寻找差异表达基因提供了一个参照。
这种方法针对无重复样本的数据是有一定优势的
目前现有的差异基因计算方法主要思路是**通过某种方法矫正后的观测基因counts代表了实际为止的真实基因counts。
**常用的矫正思路是首先考虑所含基因个数,基因表达量和样本library大小等因素进行样本间的标准化,然后使用校正后的基因counts数进行有参或者无参差异表达检验。
这些软件都有一个假设是观测到的基因counts数充分代表了真实的基因表达情,但是这一假设并不是完全没有问题。
在基因组测序中,覆盖深度是一个非常重要的概念,但是在转录组测序中因为转录组大小在不同组织或者不同细胞中都有差别,而且细胞间的mRNA种类也是高度变化的,因此覆盖深度的概念并没有很好的延伸到转录组中。依据2009年的一篇报道,想要准确覆盖人细胞系的95%转录本大致需要700M reads (但是这个说法个人认为也不能全信),因为实际测序深度远远比这个要少,所以测序带来的强大的随机效应就对统计结果有非常大的影响,目前的通用做法是尽量增加样本的重复。除此之外,现在已知 RNAseq 文库制备和测序都存在一些误差和偏好性。
从以上问题和角度出发,这篇文章提出了CORNAS(COverage-dependent RNA-Seq) 方法,利用贝叶斯方法来推断真实基因表达数的后验分布。
其创新型之一该方法包括了由RNA样品浓度决定的覆盖度参数,之二是真实基因表达量后验分布的比较为寻找差异表达基因提供了一个参照。
这种方法针对无重复样本的数据是有一定优势的,在文章中,作者也是使用的真实无重复数据进行了测试,另外比较对象也并非是DEseq和EdgeR这类有参检验的软件,而是NOISeq和GFOLD两款软件,至于使用这两款软件的原因作者提到是因为和常用的DEseq以及EdgeR相比,这两款软件返回的假阳性数目更少。
关于如何计算样本覆盖度的问题可以参考原文,另外作者发现在给定真实基因表达数时描述观测基因表达数最合适的模型是广义泊松分布(而不是过去常用的负二项分布),将真实基因数和广义泊松分布参数以及通过RNAseq样品浓度得到测序覆盖度相关联就可以确定真实基因表达量的后验分布,进而用这个分布作为无重复RNA-seq试验进行差异基因分析的基础。
综上,如果手上恰好有无重复试验的RNA-seq数据要做差异表达分析,不妨试试NOISeq,GFOLD和这个新出的CORNAS 这几个软件。
摘要
背景:
目前在RNA-Seq实验中,在调用差异表达基因的统计方法中,假设一个被调整的观察到的基因计数代表一个未知的真实基因计数。
这种调整通常包括一个归一化步骤来解释异质样本库的大小,然后将结果归一化的基因计数作为参数或非参数差异基因表达测试的输入。
一个真实基因的分布,每一个都有不同的概率,可以导致相同的观察到的基因计数。
重要的是,序列覆盖信息目前没有明确纳入用于RNA-Seq分析的任何统计模型中。
结果:
我们开发了一种快速贝叶斯方法,该方法利用RNA样本浓度确定的测序覆盖信息来估计真实基因计数的后验分布。
与NOISeq和GFOLD相比,我们的方法有更好的或比较好的性能,根据模拟实验和真实的未复制数据的实验结果。我们将先前未使用的测序覆盖参数纳入RNA-Seq数据的差异基因表达分析的过程中。
结论:我们的研究结果表明,我们的方法可用于在有限数量的复制和低测序覆盖率的实验中克服分析瓶颈。
背景
与特定表型类型显着相关的大规模的基因签名挖掘是转录组分析通常期望的结果。
RNA测序(RNA-Seq)已成为基因表达谱分析的首选工具,它在几个重要方面补充了传统的微阵列:
它更彻底地采样转录组,检测异构体,并且在没有对靶转录组的预先知识的情况下工作[1,2]。
自从发表第一篇RNA-Seq论文[3]以来,对RNA-Seq的广泛兴趣导致了测序平台如454,Illumina和Solexa的快速开发和部署。这些平台自然促成数据处理和分析方法的并行开发,以从RNA-Seq数据中提取生物学意义。
典型的RNA-Seq数据分析首先选择通过质量控制标准的读数,将它们映射到参考基因组,然后量化基因计数。标准化后,所得到的数据矩阵已准备好进行统计分析,对此可以使用令人眼花缭乱的数字替代方法[4,5]。不管这些参数是参数(例如DESeq [6],EdgeR [7],DEGSeq [8],BaySeq [9])还是非参数(例如NOISeq [10],SAMSeq [11]),观察到的基因计数是实际真实基因计数的充分表示。
在基因组测序中,总阅读长度与基因组大小的比率提供了覆盖度量,这对于评估组装基因组的完整性很重要。然而,扩展转录组大小的覆盖概念并不简单。首先,同一生物体内不同组织类型的转录组大小不同,甚至相同组织类型的细胞之间转录组大小也不同[12]。接下来,细胞之间mRNA相对比例可以变化很大[13]。例如,在基因相同的酵母细胞中,已经观察到每个细胞超过800个mRNA拷贝的变异[14]。
为了精确定量人细胞系中95%的转录物,需要高达7亿(M)的读数[15]。相比之下,RNA-Seq实验通常产生的读数远小于700M [16],足够低以使随机效应对统计分析结果的解释产生重大影响。如果不大幅度降低测序成本,研究人员往往不得不优先重复读数总数的增加,因为这是提高差异基因表达分析统计效能的最佳策略[17]。然后出现几个挑战。假设有完美的阅读图谱和定量分析,目前还不清楚观察到的基因计数是否代表真正的基因计数,因为大量的真实基因计数可能由于强大的随机效应而产生特定的观察基因计数。使问题复杂化是技术性RNA-Seq文库制备和测序中固有的偏见[18],最近才引起严重关注的问题[19]。
我们开发了CORNAS(依赖于平衡的RNA-Seq),贝叶斯方法来推断真实基因计数的后验分布。这种方法的新颖之处在于它结合了由RNA样品浓度确定的覆盖参数。随后,真实基因计数后验分布的比较为调用差异表达基因(DEG)提供了基础。我们报告了CORNAS在未复制的RNA-Seq实验中的应用,并讨论了其用于克服这些实验的分析局限性的可能性。
结果
真实基因计数和样本覆盖率的定义
我们首先将真基因计数定义为为测序运行准备的样品中基因的mRNA拷贝总数。该定义适用于包含单个或多个单元格的样本。由于后者原则上可以来源于多种不同的真实基因计数,因此不能仅凭观察到的基因计数确定该值。然而,关于样本覆盖率的信息可以改进估计真实基因计数的过程。
样品(b)的覆盖度被定义为测序的cDNA片段(S)除以总cDNA片段群体大小(N)。单端测序产生一个读数来代表一个cDNA测序,而配对末端测序产生两个读数代表一个测序的cDNA。
在Illumina测序方案中,样品覆盖率的计算可以基于mRNA样品浓度。我们推断在PCR之前的步骤中产生的cDNA的量为合理估计样本覆盖率提供了关键因素,因为:
1)样品文库制备过程中的片段化步骤导致cDNA分子大小(500bp)的同质性;
2)PCR后的体积和浓度已知(40μL的200nM cDNA)和;
3)PCR循环的数量是已知的(14个循环)。cDNA片段进行PCR以提高获得至少一个测序覆盖率的机会。
假设完美的扩增效率,每个cDNA片段在PCR过程中扩增214次。因此,PCR之前的cDNA片段数估计为4.818×1012 /214≈300M(详见附加文件1)。我们使用这个数量作为估计的片段总体大小来确定覆盖范围,因为它与我们开始使用的mRNA数量非常相似。
机会机制为观察基因计数产生广义泊松分布
当将cDNA片段加载到测序运行中时,假定从加载的cDNA片段随机产生短读数。因此,真正的基因计数会诱导观察到的基因计数的概率分布。为了找到最能描述后者的概率模型,
我们进行了一系列模拟来确定均方差观察到的基因计数在6个覆盖值下的关系:0.5,0.4,0.25,0.1,0.01和0.001。假设分别为150M,120M,75M,30M,3M和0.3M读数计算这些覆盖率从总片段群体大小300M开始测序。对于每个覆盖范围,我们生成了1到100,000的实际计数值的观察计数的经验分布(详见“方法”部分)。
模拟结果提供了三个重要观察结果:观察计数的平均值与覆盖率成正比,随着覆盖率的增加,发生不均匀分布(即方差小于平均值)(附加文件1:图S1),线性模型充分描述了平均方差比和覆盖率之间的关系(等式6)。这些结果表明广义泊松(GP)分布[20]适用于给定真实基因计数(T)的观测基因计数(X)的分布。带GP的GP的概率质量函数参数λ1和λ2由下式给出

意味着λ2= 1-√m,其中方差比(0 <m <4)。观察到的基因数的平均值,如果真实基因数与覆盖率b和真实计数k的乘积成比例,则给出λ1=bk√m,具有平均值λ1的泊松分布是当m = 1时GP的特例。
用于估计真实基因计数的贝叶斯模型观察到的基因数和测序覆盖率
公式中GP模型的重要性。 1源于反向条件使我们能够考虑的事实给出观察基因计数和测序覆盖率的真实基因计数(T)的概率分布(即真基因计数的后验分布)。让我们假设一个统一的先验分布的T值超过1,2,...。 。贝叶斯定理的应用产生:
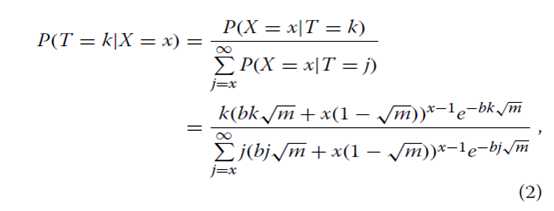
其中k≥x。请注意,尽管我们之前使用的是不正确的,但后验分布是正确的。有趣的是,伽马分布提供了一个很好的近似值等式2(数学证明见[21])。在这里,我们发现如果伽马分布的平均值μ和方差σ2与覆盖度b和观测基因数x相关,则近似值非常好(附加文件1:图S2):
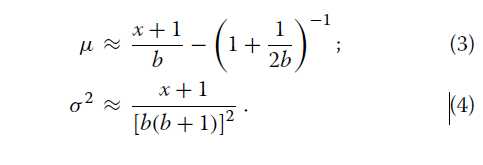
因此,近似伽玛分布的概率密度由下式给出
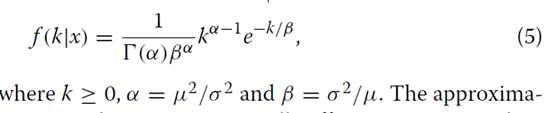
近似提供计算有效的手段来计算真实基因计数的后验分布的累积分布函数。在未复制的RNA-Seq实验中调用差异表达基因的统计检验可以基于真实基因计数的后验分布
(等式2)如下。对于单个对照和单个治疗样本,如果我们有关于测序的信息
覆盖对照样品(b0)和处理样品(b1),然后,给定观察到的基因计数
对照(x0)和治疗(x1)组,其真实基因计数的后验分布近似
伽马(公式5)。如果后者的后验平均值较大,我们宣布一个基因在治疗组中差异上调,其第0.5个百分点至少比对照组的第99.5个百分点高1.5倍(默认)。相反,如果后者具有较小的后部,则基因在治疗组中被差异下调,平均而言,其第99.5百分位至少是对照组的第0.5百分位的1.5倍(默认)(图。1)。这个过程很快,因为伽玛分布的百分位数很容易计算出来。此外,使用这个程序声明基因差异表达意味着两个样本中真实基因计数的差异为0.9952≈0.99至少1.5折。
CORNAS的性能评估
我们使用模拟和实际数据集进行了一系列比较CORNAS与NOISeq [10]和GFOLD [22]性能的测试。我们选择了GFOLD和NOISeq,因为两者都有报道在应用于未复制的RNA-Seq数据集时,与其他流行方法(如DESeq2和edgeR [5])相比,在标记为差异表达的基因中返回相对较少的假阳性数[5]。
测试1:在模拟的真实基因计数数据中检测差异表达的基因
我们使用四个覆盖范围测试了CORNAS:0.5,0.25,0.1和0.01,模拟真实基因计数范围从1到10,000。记录调用差异表达基因(DEG)的相对频率为100个独立样本
对照组与对照组之间无倍数变化(无效),1.5倍变化(弱效应)和2倍变化(更强效)的试验。在没有倍数变化的情况下,假阳性率(FPR)被估计为DEG呼叫率。真实阳性率(TPR)或灵敏度是弱和强效应情景下的DEG呼叫率。
一般来说,我们观察到随着覆盖率和覆盖率的增加,误报率降低和DEG呼叫率增加
越来越多的真实基因计数(图2)。与GFOLD和CORNAS默认值相比,当真实基因计数较低时,NOISeq产生最大的FPR。 NOISeq的灵敏度通常很好,除非0.01的低覆盖率;
当真实数量超过1,000时,其DEG呼叫率开始下降。 GFOLD表现出非常低的灵敏度,其中
与[5]中报道的保守行为是一致的。CORNAS显示出对FPR的极好控制,并且依赖于检测的倍数变化阈值DEG在弱信号和强信号情况下。例如,CORNAS默认值(φ= 1.5)在弱信号情况下表现非常差,因此如果检测到这些基因是有意义的,那么应将φ调整为较低的值,如1(CORNAS set1)。一般而言,CORNAS的灵敏度随着真实计数的增加而增加,并且覆盖值为0.1或更大时迅速收敛到1。
测试2:compcodeR模拟
观察到的基因计数的分布通常使用负二项式分布建模,并且compcodeR R软件包[23]提供了一个模拟器来模拟基于这种分布的RNA-Seq计数数据。假定基因长度相等并设定在1000个碱基处。我们使用[23]中提供的示例来创建对照组治疗比较(每组5个重复),其中对照组中有624个上调基因和625个下调基因,用于模拟12,498个基因的转录组。从这个数据矩阵中,共构建了25个未复制的数据集。对于CORNAS,我们评估了两种不同覆盖率的结果
对样本进行比较;估计比compcodeR覆盖率(CORNAS_10xless)小10倍,另一个小于100倍(CORNAS_100xless)(Supporting Data)。我们做了两个单独的NOISeq运行,一个没有长度归一化(NOISeq_nln),另一个使用M值归一化的修整均值(NOISeq_tmmnl)。
所有方法的阳性预测值(PPV)和敏感度均较低;尽管如此,CORNAS显示出比其他方法更高的灵敏度,而GFOLD具有相对更好的PPV(图3a)。所有方法的F分数非常相似(表1)。与其他方法相比,CORNAS称为更大的DEG集大小。与NOISeq_nln不同,由CORNAS调用的更大的DEG集合大小并未显着降低其PPV。CORNAS_100xless和CORNAS_10xless表现出类似的表现。
比较的平均运行时间约为NOISeq_nln和NOISeq_tmmnl的三分钟,GFOLD为一分钟,CORNAS_10xless和CORNAS_100xless为三秒。
测试3:人类性别特异性基因表达
CORNAS对真实数据适用性的评估基于人类淋巴母细胞RNA-Seq来自Pickrell的研究数据[24]。在这个数据集中,男性和女性的性别构成了两个表型类别,所以真正的DEG可以纯粹使用生物推理来确定。差异表达的基因被鉴定为具有Y染色体相关表达的19个基因[5]。在生物学基础上没有差异表达的基因包括61个X失活的(XiE)基因[25,26]和11个看家基因[27]。
我们从725对可能的组合中随机选取100对单身女性 - 单身男性对(29名女性,25名男性)(支持数据),并比较GFOLD,NOISeq和CORNAS。我们的研究结果表明,NOISeq与CORNAS和GFOLD相比表现不佳,而GFOLD表现略好于CORNAS(图3b,表1)。但是,类似于compcodeR仿真结果,CORNAS称为较大的DEG集。
NOISeq的平均运行时间约为2分钟,GFOLD为30秒,CORNAS为10秒。
测试4:在组织特异性基因表达数据中的覆盖效应
Marioni数据集[28]由来自人类肝脏和肾脏的RNA-Seq数据在两种不同的序列中组成加载浓度,3 pM(高)和1.5 pM(低)。我们调查了CORNAS是否会被简单地根据不同浓度进行DEG调用时被误导,当两个样品都来自同一组织时。在CORNAS中假阳性率低,在相同浓度的相同组织样品中没有DEG呼叫进行比较(附加文件1:表S1)。但是,对于不同的样品GFOLD显示出比CORNAS更少的误报。在所有情况下,NOISeq都返回了最高FPR。
一组4863个基因被鉴定为在组织表达数据库TISSUES中编目的人肝脏或肾脏组织中唯一表达[29]。同样,与CORNAS和GFOLD相比,NOISeq表现不佳,而CORNAS表现最好(图3c,表1)。对于不同组织类型之间的所有12种比较,最大的DEG组被CORNAS调用,并且NOISeq最小的那个。
通常对于不同的组织类型,由NOISeq和GFOLD调用的DEG集显示差的重叠,与GFOLD和CORNAS之间的重叠相比,和NOISeq和CORNAS之间(附加文件1:图S3)。CORNAS表示针对不同组织类型的更独特的DEG呼叫。与此同时,来自GFOLD或NOISeq的大部分DEG呼叫也被CORNAS调用。
NOISeq的平均运行时间约为5分钟,GFOLD约为30秒,CORNAS为5秒。
PCR扩增效率对灵敏度的影响
虽然我们在构建模型时假设了完美的PCR扩增效率,但我们仍在评估可能性影响95%,90%,85%和80%PCR效率对CORNAS灵敏度和FPR的影响(详见“方法”一节)。正如我们没有的那样,CORNAS似乎对小的违反完美PCR扩增效率的鲁棒性,即使PCR效率达到80%,也会发现灵敏度和FPR的实质性变化。曲线下面积(AUC)为所有四种测试预期覆盖率的接收者操作特征(ROC)图均小于5%(图4和附加文件1:图S4)。
讨论
CORNAS作为估计真实基因数量的框架
作为RNA-Seq计数数据建模中负二项分布的替代方案,GP模型正在被越来越多地研究[30-33]。在这里,我们展示了一种机会机制,它自然使GP成为观察基因计数数据的模型。通过将GP的参数与真实基因计数和测序联系起来使用RNA样本浓度的覆盖率,我们因此能够确定真实基因计数的后验分布。这种分布形成了在未复制的RNA-Seq实验中进行DEG调用的基础。
目前,在生物体的基因模型上映射的读取深度用于估计RNA-Seq实验中的覆盖度。我们知道样品中mRNA的总量在Illumina测序仪中未被捕获,其具有固定的有限饱和量,其可能超过或低于样品浓度。覆盖面通常被认为是代表性不足,这是一种限制这通常被认为是可以通过深度测序纠正的,该测序用于检测mRNA表达非常低的基因[15,17,34]。我们覆盖参数的范围(0到1之间)应该覆盖最多没有完成深度排序的实际案例。如果估计的覆盖范围不止一个,我们不建议使用CORNAS。
我们的研究做了几个假设来简化模型,其中之一是100%有效的PCR扩增。我们模拟的PCR扩增效率的影响确实表明,当我们高估覆盖率时,灵敏度和FPR增加,但是这种变化没有不利影响。为了保持观察计数模型,在当前工作中假设理想的随机cDNA片段采样(因此后验分布)我们可以非常简单地研究将覆盖参数引入到DEG呼叫过程中的效果。由于真正的RNA-Seq实验含有文库制备偏差,通过完整的测序过程模拟器,如rlsim [35],可以更好地探索这种偏差的影响。
在我们的模拟中未明确处理观察到的基因计数的潜在变异来源,涉及不同算法将短读数映射到参考基因组的方式(例如使用BWA [36],OSA [37],TopHat [38]和Bowtie [39]),以及如何量化这些映射读数(例如使用HTSeq [40]和Cufflinks [38])。我们建议由于这种变异来源观察到的基因计数的变化是相对的不重要,因此不会严重影响真实基因计数的后验分布。首先,可以提高读取对齐质量的算法[41],从而最大限度地减少计数错误。此外,read-mapper和基因计数量化的组合已经经过实验研究,并且可获得最可靠的最佳建议以获得最可靠的观察到的基因计数(如[42]所建议的OSA + HTSeq)。
CORNAS的稳健性
尽管如此,CORNAS在compcodeR仿真中表现出与GFOLD和NOISeq相当的性能,基于观察到的基因计数的不同数据模型(即广义泊松比负二项式)。该发现提供了将CORNAS框架整合到当前RNA-Seq数据分析方案中的信心。此外,尽管覆盖范围已被估计,并因此出现错误,但两个CORNAS设置(10xless和100xless)平均显示类似的性能。CORNAS达成了一个很好的妥协,与GFOLD和NOISeq相比,灵敏度,PPV和DEG设置大小之间。在现实世界的实验中,CORNAS如果能够更可靠地确定覆盖范围,则可以超越竞争方法,例如来自Marioni数据集在测试4中。
如果不包含coverage参数中的信息,诸如GFOLD和NOISeq等传统方法用于分析未复制的RNA-Seq计数数据要么过于保守,要么很少打电话,但其中大多数是真正的阳性(GFOLD),或者在非常低的覆盖范围情况下(例如b = 0.01)做出相对更多的误报(NOISeq)(图2)。另一方面,我们发现CORNAS很好地控制了FPR并且当覆盖率不太小时(例如b≥0.1)具有高TPR。此外,如果检测到较弱的倍数变化差异是令人感兴趣的,
那么可以将折变参数(φ)从1.5减小到1.0(详见“方法”部分)。对于弱信号和强信号,CORNAS在倍数变化参数为1.0时的TPR分布与NOISeq类似,除非覆盖率非常低。随着真实基因数量的增加,CORNAS继续显示TPR普遍上升,而NOISeq则下降。
现在,大多数RNA-Seq实验在测序前没有报告起始材料中实际RNA量的估计值。因此,我们只能通过模拟研究使用后验均值校正观察基因数的效果。鉴于令人鼓舞的结果,研究人员可能希望收集关于未来覆盖参数的信息,以利用CORNAS分析真实RNA-Seq数据集。
分析未复制的RNA-Seq计数数据时的一个主要问题是在缺少生物学重复时缺乏有效的归一化方法。在这里,我们已经展示了CORNAS所基于的贝叶斯框架,通过处理基因真实计数的后验分布来避免归一化问题。结果,转录物长度信息不是必需的。这使得CORNAS适用于具有不完整或不断演变的转录组参考数据的生物体,因为新的转录信息不会改变真实计数随着时间的推移如何估计。我们的研究结果表明,CORNAS可以用作克服分析瓶颈的手段,有限的重复和低测序覆盖率导致DEGs,使用定量PCR和NanoString nCounter [43]等平台进行下游验证具有更好的前景。将CORNAS扩展到多重复制的结果将在其他地方发布。
结论
我们开发了CORNAS(COverage-dependent RNA-Seq),一种快速贝叶斯方法,它结合了一个新的覆盖参数来估计真实基因计数的后验分布。在CORNAS框架下,可以使用由RNA样品浓度确定的来自序列覆盖的正交信息来提高调用DEG的准确性。通过对实际数据集的模拟和分析,我们发现在未复制的RNA-Seq实验的情况下,CORNAS的性能与GFOLD和NOIseq相当或更好。
方法
CORNAS作为R程序实施,可供下载(https://github.com/joel-lzb/CORNAS)。用于模拟和数据分析工作的Perl和R脚本可在https://github.com/joel-lzb/CORNAS_上找到Supporting_Data。我们在运行RedHat 6操作系统的96 GB RAM的IBM System x3650 M3(2x6核心Xeon 5600)机器上进行了计算机实验。图形使用R [44]中的ggplot绘制。模拟片段抽样过程以及覆盖率和观测计数的均值/方差之间的关系,考虑具有相同长度的N个cDNA片段的群体。在这项研究中,我们设置N = 300×106(300M)。我们使用以下数目的测序读数(S):150M,120M,75M,30M,3M和0.3M,用于模拟0.5,0.4,0.25,0.1,0.01和0.001的覆盖率(S / N)分别。
为了模拟来自cDNA片段群体的取样过程,我们首先对每个N cDNA进行索引分子从1到N.接下来,我们使用Fisher-Yates shuffle算法来洗牌指数,创建置换π=(π1,π2,...,πN)。π的前S个元素表示测序片段的索引。对于从1到100,000的每个真实基因计数k,我们确定相应的观察基因数为

其中IA是事件A为真时的值为1的指示器函数,否则为0。总共进行2000次迭代,并从中估计观察到的基因计数的平均值和方差。理论上,从这个过程中产生的观察计数遵循超几何分布。因此我们能够计算给定覆盖率b的超几何分布的均值和方差(m)的比率为:
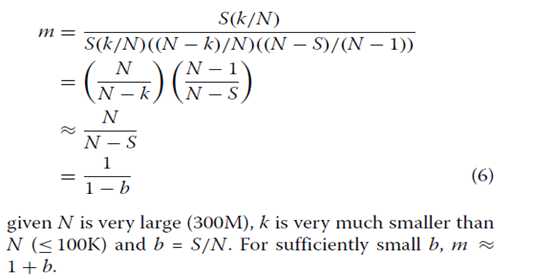
如果N非常大(300M),k远小于N(≤100K),b = S / N。对于足够小的b,m≈1+ b。
抽样过程自然导致观测计数的超几何分布,因为N是有限的。然而,实际上N很大并且
未知,因此需要一个没有上限的近似分布(参见GP模型中的“结果”部分)。
将后验均值和后验变化建模为覆盖的函数
观察计数x的真实计数k的后验分布如公式2。然后我们确定了后验分布的均值和方差与覆盖参数的关系。我们首先将均值和方差建模为线性函数,使得:
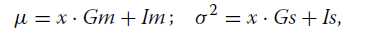
其中参数Gm和Gs是梯度,并且Im和Is分别是截距。然后,我们为每个参数拟合模型
在各种覆盖范围进行模拟(附加文件1:图S2)。等式3和4是模拟均值的最终近似值和作为观察基因计数(x)和测序范围(b)的函数的后验分布的变化。
CORNAS评估
程序设置
需要在CORNAS中设置两个参数。第一个是α,其用于确定较低(1-α)/ 2×100百分位数(p(1-α)/ 2)和上(1 +α)/ 2×100百分位数(p(1 +α)/ 2)。第二个参数是折变截止φ。为了进行DEG调用,我们需要p +(1-α)/ 2 / p-(1 +α)/2≥φ,其中上标+和 - 分别表示后验分布具有较高和较低的平均值。默认设置是α= 0.99和φ= 1.5。这些值可以改变CORNAS更保守(例如增加α和/或φ)或更自由(例如降低α和/或φ)。NOISeq在q = 0.9截止时运行。 GFOLD运行的折叠变化为0.01。如果基因的表达被认为是上调的GFOLD值为1或更大并且如果GFOLD值为-1或更小则下调
性能指标
真阳性(TP)是已知在两个样品之间差异表达的基因,并且通过评估的方法检测为DEG。假DEG电话是误报(FP),而假阴性(FN)则错过了真正的DEG呼叫。对于DEG调用方法,其正向预测值(PPV)是正确调用的比例DEG(TP /(TP + FP)),其灵敏度是称为真实DEG的比例(TP /(TP + FN))。我们共同考虑了测试2,3和4的每种方法的灵敏度和PPV。计算每个比较的F评分(灵敏度和PPV的调和平均值)为2×(灵敏度×PPV)/(灵敏度+ PPV)??。报道了每种方法的Themean F评分。
对于测试1,假阳性率(FPR)根据无倍数变化情况确定为真阴性(TN)明确已知
(FP /(FP + TN)),而灵敏度的计算方法与弱,强效应场景中的测试2,3和4相似。
测试1:在模拟的真实基因计数数据中检测差异表达的基因
对于此模拟,考虑了三种生物效应情景:无倍数变化(无效),1.5倍变化(弱效果),2倍变化(强效果)。在这三种情况下考虑的最大真实数分别为10,000,6,666和5,000。
我们假定每个真实的基因计数都是由一个基因发出的,因此在所有三种情况下所有真实基因计数的集合总共对应于21,666个基因。按照模拟片段抽样过程中描述的程序产生每个基因的观察计数。总共进行了100次迭代说明观察到的基因计数的取样变异性。在特定方法需要基因长度信息的情况下,我们将其设置为1000个碱基。
测试2:compcodeR模拟
我们根据compcodeR论文[23]中描述的方法生成模拟数据集B_625_625。DEG的数量占基因总数的10%(12,498)。
测试3:人类性别特异性基因表达
对于来自尼日利亚的29名女性和25名男性组成的Pickrell研究,我们使用了总数
来自已发表论文[24]的测序读数和来自ReCount数据库的RNA-Seq计数数据[45]。每个样品的测序覆盖率计算为报告的总读数除以标准300M cDNA片段大小。对于具有多个测序运行的样品,我们取所生成的总读数的平均值。
测试4:在组织特异性基因表达数据中的覆盖效应
在Marioni数据集中,相同的人类肝脏和肾脏样品在每条七条泳道中测序,其中5个泳道的RNA浓度为3pM,另外两个泳道的浓度为1.5pM。14条泳道分两次进行测序。为了减少技术变化,我们仅使用来自运行2的数据,其中不同浓度的加载在相同条件下运行和时间。我们将代表样品转录组的cDNA片段的数量估计为负载浓度的乘积,加载量(假定标准为120μL)和Avogadro常数6.022×1023mol-1。根据2016年6月14日提取自TISSUES [29]的策划信息确定了所使用的真实的DEGs集。我们选择了737个人类肾脏基因和4,126个人类肝脏基因,这些基因支持实验验证结果,并且可以用Ensembl基因ID鉴定。
PCR扩增效率对灵敏度的影响
使用与测试1相同的数据集进行评估。为了模拟研究中PCR扩增效率的影响,
我们重新计算了每种COSNAS的测序覆盖率,通过减少由PCR不同扩增效率引起的PCR之前假定的片段总数(分别为95%,90%,85%,80%扩增效率的总片段分别为70%,49%,34%,23%)。有关如何确定PCR扩增效率的详细信息可以在附加文件1中找到。
例如,有一个样本完全扩增,但测序范围为0.25,在PCR之前将有300M片段,并产生75M读数。假设产生的读数保持不变,但PCR扩增效率现在为95%,则测序覆盖率估计将为0.36(75M /(300M×0.7))。新的覆盖范围然后用于0.25覆盖的CORNAS以95%的PCR扩增效率运行。对于每个覆盖范围,根据在无效情况下调用的DEG的数量计算FPR,
并且从强效果方案中调用DEG计算灵敏度。我们使用ROCR R软件包[46]生成了接收器操作特性(ROC)曲线。通过固定α= 0.99然后从1.5变化到0.75获得用于制作差异表达呼叫的截止点,并通过固定φ= 0.75,然后将α从0.99变为0.01。
以上是关于无生物学重复RNA-seq分析 CORNAS: coverage-dependent RNA-Seq analysis of gene expression data without biologi的主要内容,如果未能解决你的问题,请参考以下文章