易基因|单细胞转录组测序:Smart-seq2和10X Genomics Chromium怎么选?
Posted E-GENE
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了易基因|单细胞转录组测序:Smart-seq2和10X Genomics Chromium怎么选?相关的知识,希望对你有一定的参考价值。
应用单细胞测序技术研究科学问题越来越普遍,当下应用最火热的是10X Genomics公司的Chromium解决方案(以下简称10X)。但是基于2017年Christoph Ziegenhain et al[1]对6种单细胞转录组技术及2019 Broad研究所的团队对7种单细胞RNA测序方法进行了比较[2],某些特殊或者少量细胞样本的单细胞转录组研究中,Smart-seq2技术还是一项研究利器。
Smart-seq2与10X技术的区别与应用的科研场景有哪些呢?北京大学张泽民团队在2019年发表的预印文章“Direct Comparative Analysis of 10X Genomics Chromium and Smart-seq2”中给予了答案。
那么今天小编再次对Smart-seq 2与10X Genomics Chromium的技术原理和应用进行阐明。
Smart-Seq 简介
Smart-Seq (Switching mechanism at 5\' end of the RNA transcript)于2012年发表[3],2013年发表了其改进技术的应用Smart-Seq2[4],2014年Smart-Seq2 protocol发表[5]。Smart-Seq2 对原始的Smart-Seq实验流程进行了多项改进优化,它不再需要纯化步骤,可大大提高产量,最重要的改进是下面两项:
-
TSO 3\'端最后一个鸟苷酸替换为锁核酸LNA(locked nucleic acid)。LNA单体的热稳定性增强,其退火温度增强非模板cDNA的3\'延伸能力。
-
甜菜碱(一种具有两个重要作用的甲基供体:它会增加蛋白质的热稳定性,并通过破坏DNA螺旋来降低甚至消除了DNA热融变对碱基对组成的依赖性)与较高的MgCl2浓度结合使用。解决某些RNA形成二级结构(例如发夹或环)由于空间位阻,可能导致酶终止链延长的问题。
Smart技术是基于高保真的反转录酶、模板转换和前置放大来增加cDNA得率,实验流程2天,得到的是全长转录本。该方法有较好的覆盖范围,可检测到稀有转录本,不需要额外的专业设备,因此应用范围较广。
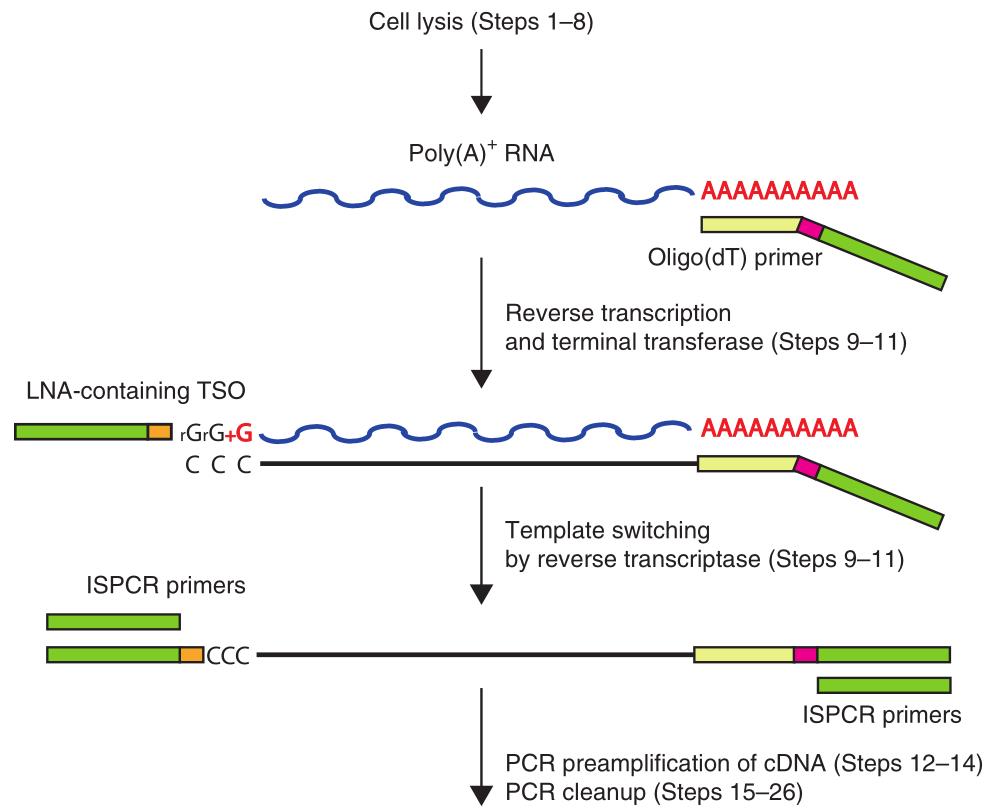
Smart-Seq2建库原理
-
1. 单细胞分选:Smart-seq2使用流式细胞仪或显微操作进行细胞分选,体积不超过0.5 ul。
-
2. 细胞裂解:将分离细胞直接转移到细胞裂解液中进行细胞裂解。
-
3. 反转录( 一链合成 ):使用Oligo(dT) primer 对带有polyA尾的RNA( 主要mRNA )进行反转录。由于使用了特殊活性反转录酶( Moloney Murine Leukemia Virus )进行反转录,所以会在cDNA链3\'端加上三个C。
-
4. 模板置换( 二链合成 ):该步使用TSO ( template-switching oligo )引物合成了cDNA的二链,从而置换了与一链cDNA互补的RNA。要注意的是TSO 3\'端有三个G能与一链3\'端的三个C互补,而最末端的+G是一个修饰过的G,能增加TSO的热稳定性,以及其与一链cDNA游离的3’端的互补的能力。
-
5. PCR扩增:该步进行轻度的cDNA富集,将cDNA扩增至ng级即可。
-
6. 标记:利用改造后的高活性Tn5转座酶对DNA进行打断的同时将接头添加到cDNA的两端。标记完成后的DNA片段通常在200-600bp。
-
7. PCR富集及上机测序:在进行最后一次PCR扩增后,即可上机测序。
10X Genomics单细胞技术
ChromiumTM Single Cell 3\' Solution是基于10X Genomics平台,能够一次性分离、并标记5000–10000个单细胞,并能在单细胞水平进行检测的技术。该方法基于微流控技术(Microfluidics-based approaches),与Smart有相似的分子生物学原理,运用了模板转换技术,但与Smart的细胞捕获和通量不同。droplet-based方法是将单个细胞包裹在一个小油滴中(含有barcode和RT primer)反转录成cDNA,然后油滴破裂释放cDNA,统一进行文库构建,增大了实验通量,但需要专门的实验设备。
10X Genomics建库原理
-
1. 制备好的细胞悬液、10X barcode凝胶磁珠和油滴分别加入到Chromium Chip B的不同小室,经由微流体“双十字”交叉系统形成GEM。为了获得单细胞反应体系,细胞悬液浓度建议控制在700-1200细胞/ul,因此产生的90-99% GEM不含有细胞,剩下的大部分GEM含有一个细胞。
- 2. 单个GEM依次形成后再全部混合,细胞裂解,凝胶珠自动溶解释放大量引物序列。

3. 释放的引物中包含30nt poly dT反转录引物,带有polyA的RNA被反转录为带有10X Barcode和UMI信息的cDNA一链,再以SMART方式完成二链合成。
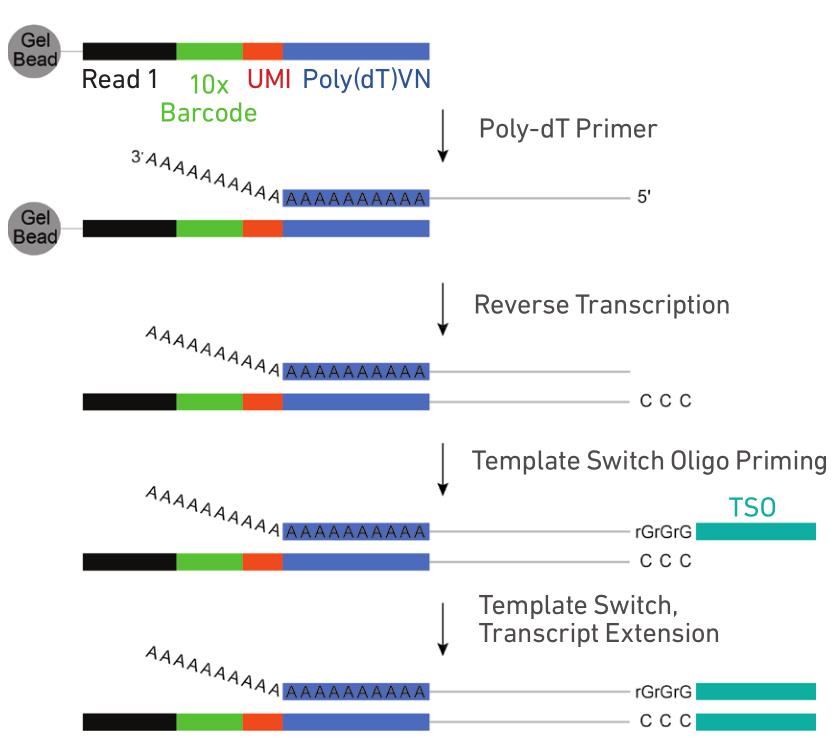
4. 油滴破碎,磁珠纯化cDNA一链,然后PCR扩增cDNA。
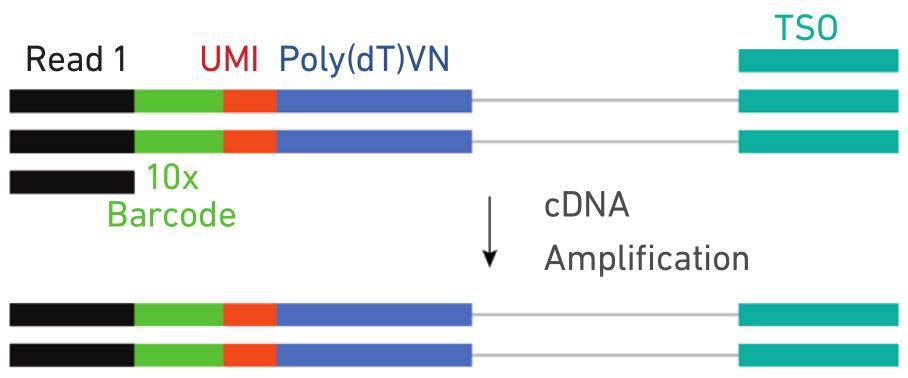
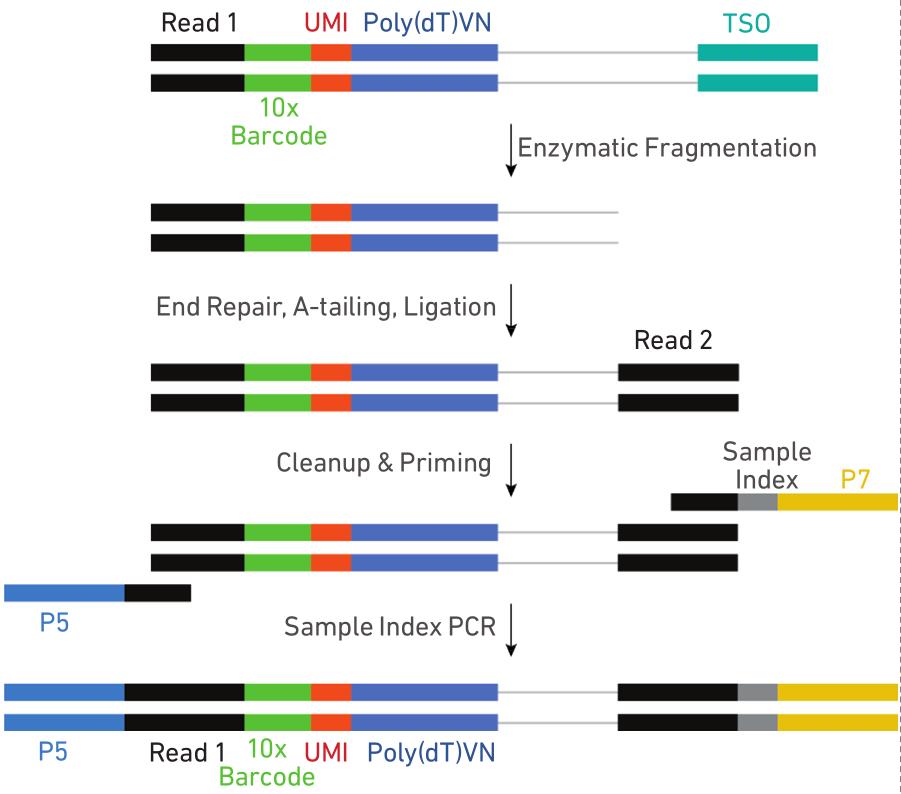
5. cDNA扩增完成后酶切片段化并磁珠筛选最适片段,通过末端修复、加A、接头连接Read2测序引物,再以PCR方式构建含有P5和P7接头的cDNA文库即可。
Smart-Seq2 和 10X Genomics
优点与限制

数据差异
张泽民团队通过直接比较两个平台上来自相同CD45细胞样本的scRNA-seq数据,广泛系统地分析评估了各自数据特征。Smart-seq2在细胞中检测到更多的基因和更为复杂的数据集,尤其是丰度较低的转录本以及可变剪切的转录本,但捕获了更高比例的线粒体基因。Smart-seq2数据的组合也更类似于bulk RNA-seq数据。

尽管都是基于poly(A)富集策略,两个平台检测到的所有转录本中约有10-30%来自非编码基因,而lncRNA在10X中所占的比例更高。
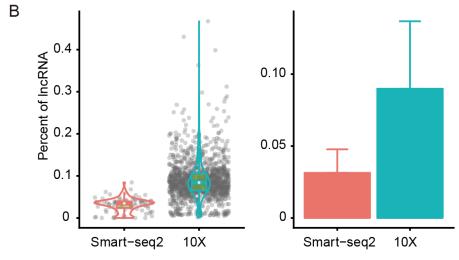
Smart-seq2具有更高的敏感度,检测到的表达基因数目远高于10X的检测到的表达基因数目。

对于检测到的基因,Smart-seq2数据呈单峰分布,在所有细胞中检测到的低表达基因很少。相比之下,10X的数据由于有大量的接近零表达的基因,呈现出明显的双峰分布,说明10X数据中mRNA在很低的表达水平上有较高的噪声或随机捕获。

基于10X的数据显示出更严重的dropout问题(dropout是scRNA-Seq数据的一大特点,就是很多基因在某些细胞根本就检测不到表达,但是在另外的细胞却检测为高表达。通常认为这个dropout是因为在文库构建的过程中,有部分基因没有被成功的反转录。),尤其是对于表达水平较低的基因。

10X 技术测序数据由于技术特色在mRNA的3’端有着较强的bias,而Smart-seq2技术则在gene上有着更为均一的分布。同时, 10X技术数据在splicing分析层面并不适用。
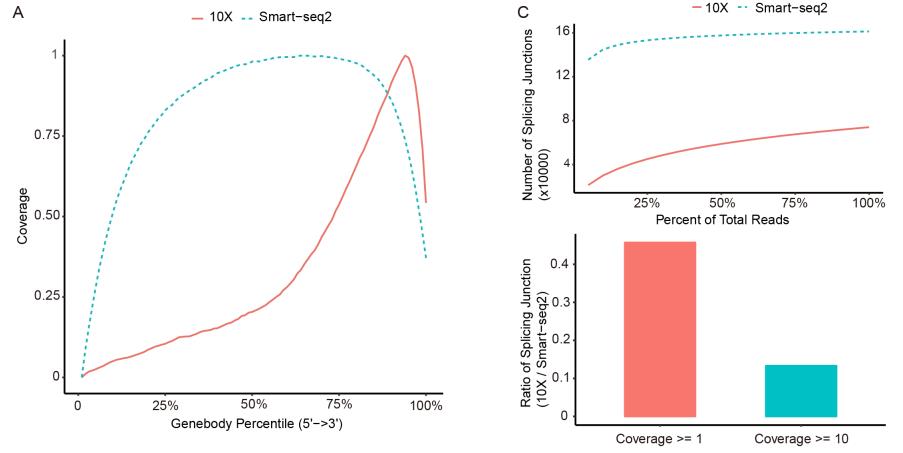
但是,由于10X数据能够覆盖大量细胞,因此可以更好地检测稀有细胞类型。此外,每个平台检测到的细胞簇之间差异表达基因的不同集合,表明这些技术是具有互补性的。
Smart-seq2技术案例
Björklund 2016年利用Smart-seq2单细胞转录组测序揭示人CD127+先天淋巴细胞的异质性[6]。先天淋巴细胞(ILCs)是一种免疫细胞,它通过释放可溶性的因素调节对病毒、细菌和寄生虫的早期免疫反应。这些细胞可以根据细胞表面的转录因子及细胞因子被分为三个亚型,每种亚型都有不同的功能。利用流式细胞仪分选患有阻塞性睡眠呼吸暂停综合症患者的扁桃体组织中的ILCs(Lin−CD127+)和NK cells(CD45+Lin−CD127−NKG2A+CD56+CD16− )。采用Smart-seq2进行单细胞转录组扩增建库;Illumina HiSeq 2000 3M reads /样本进行测序分析。采用PCA(主成分分析)和t-SNE对ILC中847个表达的基因进行分型分析,将细胞分为4类:ILC1、ILC2、ILC3、NK cells。应用SCDE软件包对4类细胞的差异基因进行分析,找出共有及特有差异基因,结果发现,CD127+ILCs中共有的差异基因的表达量比NK细胞中明显要高。ILC1、 ILC2、ILC3差异基因表达分析,结果表明,ILC1 cells共有79个上调表达基因,参与干扰素γ的调控,ILC2 cells共有58个上调表达的基因,在前列腺素、Notch信号通路及环境感应中发挥作用,ILC3 cells共有371个上调表达的基因,根据GO注释有85个与免疫相关,且存在未知功能的基因。
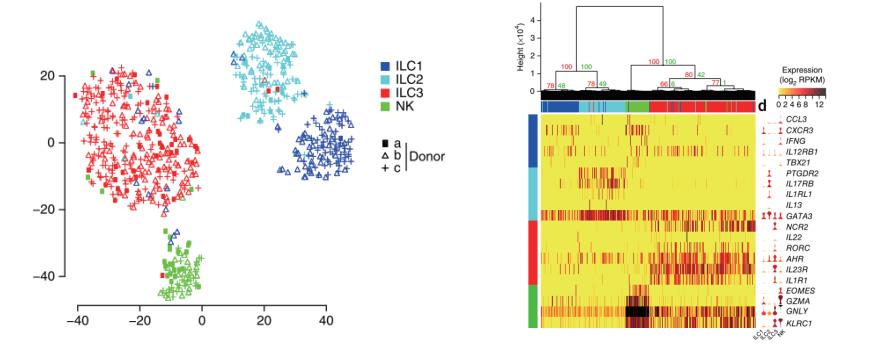
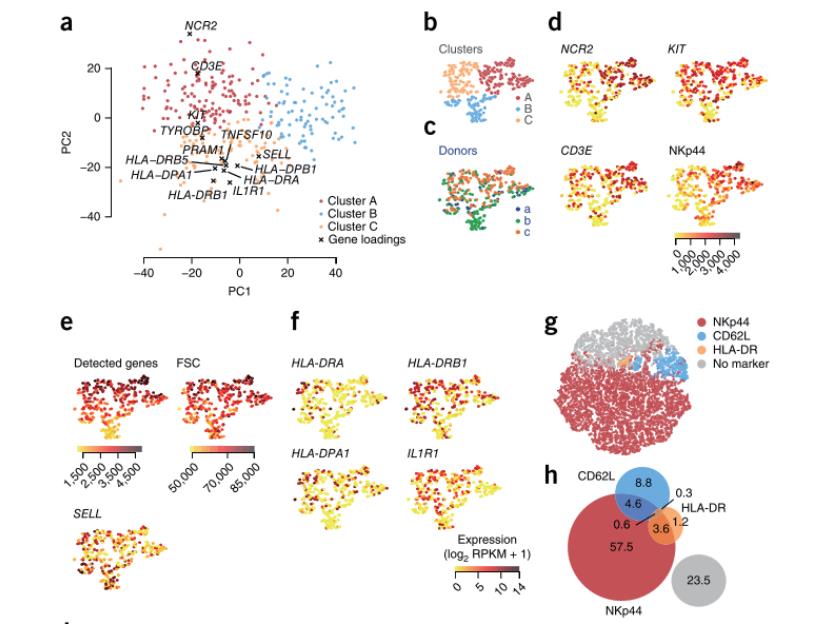
采用PCA和t-SNE分析ILC3中1,958个注释的免疫基因,将ILC3分为3个亚型:Cluster A、Cluster B、Cluster C, 通过对这3种亚型细胞的分析,发现了新的免疫细胞CD62L+ ILC3。本研究通过对648个单细胞进行单细胞转录组的分析,应用t-SNE细胞分型、SCDE基因差异表达分析,在ILC3中发现新的免疫细胞CD62L+ ILC3。
10X Genomics技术案例
比利时的研究团队通过对数以千计的健康与癌变肺细胞的研究,创建了史上第一个完整的肺癌细胞图谱。研究结果表明,肺癌较先前认识复杂得多,其包含了52种不同类型的基质细胞,这些新信息可用于开发新的肺癌治疗途径[7]。
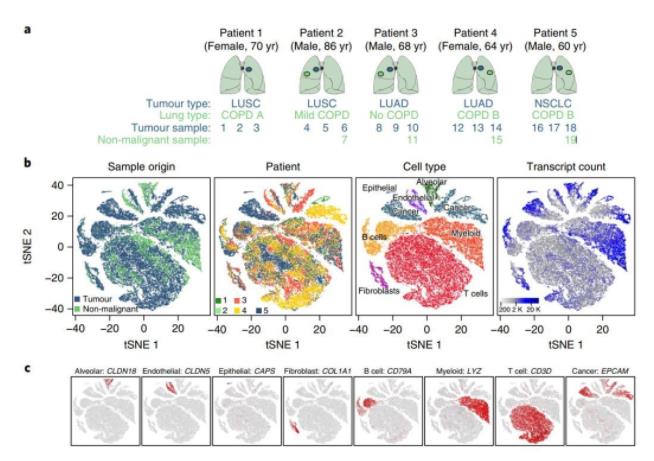
Smart-seq2与10X Genomics联合技术案例
2019年10月31日,北京大学生物医学前沿创新中心(BIOPIC)、生命科学学院、北京未来基因诊断高精尖创新中心(ICG)张泽民教授、任仙文副教授联合首都医科大学附属北京世纪坛医院彭吉润教授以及勃林格殷格翰公司刘康博士,在Cell上发表了题为Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma的研究论文[8]。该研究结合10x Genomics和SMART-seq2两种单细胞RNA测序技术,对肝癌患者多个组织的免疫细胞做出了系统性的刻画,分析了免疫细胞动态迁移和状态转化的特征,探索了它们在肝癌治疗上的潜在价值。
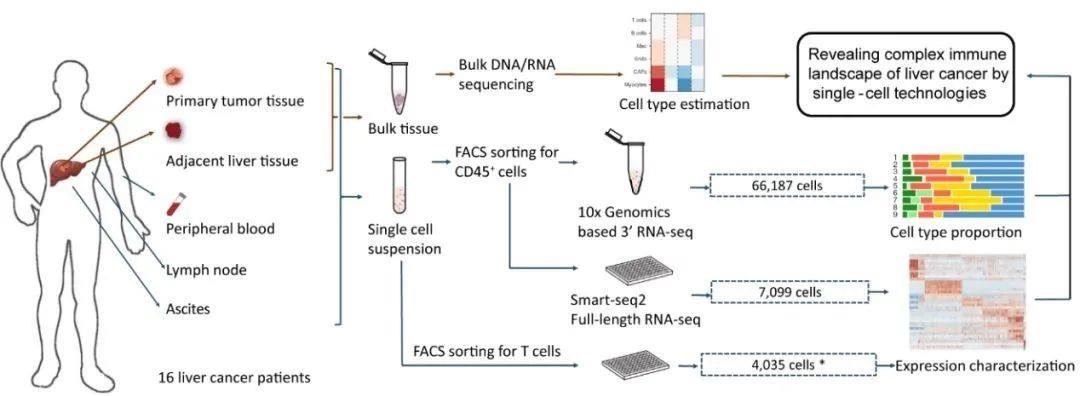
主要发现
-
不同组织的免疫组成有较大差异,肿瘤中的巨噬细胞构成腹水中髓系细胞的主要来源。
-
肿瘤中的巨噬细胞呈现两种不同的状态(TAM-like和MDSC-like)通过与其他数据中3. 肿瘤中的LAMP3+ DC是成熟态的DC,具有向肝淋巴结迁移和与多种淋巴细胞相互作用的潜在能力。
参考文献:
-
Ziegenhain, C., et al., Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol Cell, 2017. 65(4): p. 631-643 e4.
-
Vieth, B., et al., A systematic evaluation of single cell RNA-seq analysis pipelines. Nat Commun, 2019. 10(1): p. 4667.
-
Ramskold, D., et al., Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol, 2012. 30(8): p. 777-82.
-
Picelli, S., et al., Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods, 2013. 10(11): p. 1096-8.
-
Picelli, S., et al., Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc, 2014. 9(1): p. 171-81.
-
Bjorklund, A.K., et al., The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol, 2016. 17(4): p. 451-60.
-
Lambrechts, D., et al., Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med, 2018. 24(8): p. 1277-1289.
-
Zhang, Q., et al., Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell, 2019. 179(4): p. 829-845 e20.
关于易基因单细胞转录组测序
(smart-seq2)
时下火热的10X Genomics公司Chromium解决方案无法满足某些特殊或者少量细胞样本甚至单细胞转录组研究。Smart-seq2技术在单细胞水平对带Poly(A)的RNA进行全长转录组扩增及高通量测序,能够满足高灵敏度、低偏好性的cDNA扩增,得到全长转录本,实现高水平的序列模板转换。
Smart-seq2技术有较好的覆盖范围,可检测到稀有转录本,无需额外的专业设备,应用范围较广,可以解决传统 RNA 定量技术在早期胚胎发育、干细胞、癌症、免疫等研究领域中存在的样品量极低或细胞异质性的问题,是在单细胞水平研究基因表达强有力的工具,极大地拓展了RNA-seq 的应用范围。
技术优势:
起始量低:1-1000个细胞或10pg-10ng total RNA即可高效扩增;
转录本覆盖度高:通过双端引物扩增全长cDNA,获得全转录组信息,避免3’端和5’端偏好性,产物完整性好;
检测灵敏度高:大幅度增加了低表达基因的检出量;
碱基分辨率高:可达单碱基分辨率,且可以定位到具体基因的具体转录本;
实验可控:质控点多,可从实验的开端判断细胞状况。
10X单细胞转录组整合、转录组 && ATAC整合分析之VIPCCA
参考技术A单细胞测序在基因调控、细胞分化和细胞多样性研究中具有革命性意义 。 随着近年来技术的显着改进,每个实验检测的单细胞数量呈指数级增长,同时大规模研究产生的数据集也在快速增长和积累。 因此, 当前单细胞研究中的一个主要计算挑战是对来自多个不同样本或跨不同平台和数据类型的测量进行标准化,以进行有效的综合和比较分析 。 这种综合分析需要开发单细胞数据对齐方法,该方法可以消除批次效应并考虑跨数据集的技术噪声。
最近开发了许多单细胞数据对齐方法 。它们中的大多数,除了一些值得注意的例外,例如最近的 iNMF ,都针对小型和中型数据集。这些现有的方法可以概括为四类:(i) 基于参考的方法 ,例如 Scmap-cluster 和 scAlign,它们基于注释良好的参考数据集对齐新的查询数据集; (ii) 基于聚类的方法 ,例如 Harmony 、DESC,它们通过迭代优化聚类目标函数来消除批效应并在嵌入空间中对齐样本; (iii) 基于匹配的方法 ,例如 MNN 和 Scanorama ,它们应用相互最近的邻居策略来识别跨数据集的重叠单元格和 (iv) 基于投影的方法 ,使用统计模型将来自不同数据集的单个细胞投影到较低的维空间,包括对投影应用典型相关分析的 Seurat ,使用来自非负矩阵分解的潜在因子进行投影的 LIGER , and scVI and others that use variational techniques for projection.
然而, 大多数现有的对齐方法都存在固有缺陷,无法成功应用于大型数据集 。具体而言, 基于参考的方法的对齐将受到参考数据大小和参考中可用的预选细胞类型注释的限制,因此当数据大小增加时,可能会导致错过新发现的机会增加 。像 MNN 这样的基于匹配的方法使用往返游走策略,该策略需要为具有两个以上样本的数据集生成所有成对对齐,这对于大样本量来说将是耗时的。具有复杂参数模型的方法(例如 LIGER 和 scAlign)或具有复杂事后数据处理的方法(例如 Seurat )也难以扩展到大型数据集。 基于 ZINB 的方法(例如 scVI)在捕获多个数据集的复杂表达特征方面可能效率较低 。尽管一些现有的最新方法可以扩展到大型数据集,但由于复杂的参数模型,它们仍然有可能不准确地对齐细胞。因此, 迫切需要开发在计算上也有效的有效对齐方法 。
除了迫切需要开发可扩展的比对方法外,当前比对方法的另一个阻碍问题是它们的性能通常仅使用单细胞 RNA 测序 (scRNA-seq) 数据进行基准测试和优化。 因此,大多数现有的比对方法不适合整合其他单细胞测序数据类型,例如使用测序 (scATAC-seq) 进行转座酶可及染色质的单细胞测定。 此外, 现有的比对方法(如 Seurat)返回的结果只能保留真实的细胞间关系(或相似性),而不能代表基因表达水平,不适合进行差异表达分析或富集分析等下游分析 。
为了应对这些挑战, 作者提出了一个统一的计算框架 VIPCCA,它基于非线性概率典型相关分析,用于有效且可扩展的单细胞数据对齐 。 VIPCCA 利用来自深度神经网络的尖端技术对单细胞数据进行非线性建模,从而允许用户通过跨技术、数据类型、条件和模式的多个单细胞数据集的集成来捕获复杂的生物结构。此外,VIPCCA 依靠 变分推理 来进行可扩展计算,从而能够将大规模单细胞数据集与数百万个细胞有效集成。重要的是,VIPCCA 可以将多模态转换为低维空间,而无需任何事后数据处理,这是与现有对齐方法形成直接对比的独特且理想的功能。
加载
Loading data
该函数仅适用于 fit_integrate() 函数训练生成的 AnnData。 在基因表达矩阵中随机选择 2000 个位置。 x轴代表这些位置原始数据的表达值,y轴代表同一位置的vipcca整合后数据的表达值。
After filter, we converting Seurat Object to AnnData via h5Seurat using R packages. In this case, the atac.h5ad file will be generated in the corresponding path .
生活很好,有你更好
以上是关于易基因|单细胞转录组测序:Smart-seq2和10X Genomics Chromium怎么选?的主要内容,如果未能解决你的问题,请参考以下文章