易基因: oxRRBS+RRBS揭示炎症性肠病导致发育异常的表观遗传机制|甲基化研究
Posted E-GENE
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了易基因: oxRRBS+RRBS揭示炎症性肠病导致发育异常的表观遗传机制|甲基化研究相关的知识,希望对你有一定的参考价值。
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
2020年12月31日,美国明尼苏达大学Natalia Y. Tretyakova教授团队在《Int J Mol Sci》杂志发表题为“Multi-Omics Characterization of Inflammatory Bowel Disease-Induced Hyperplasia/Dysplasia in the Rag2-/-/Il10-/- Mouse Model”的研究文章,该研究通过简化基因组甲基化测序(RRBS)+氧化简化基因组甲基化测序(oxRRBS)及对应的转录组测序(RNA-seq)揭示炎症性肠病 (IBD)导致基因组中甲基化和羟甲基化模式变化,表明了IBD导致发育异常的表观遗传机制。

标题:Multi-Omics Characterization of Inflammatory Bowel Disease-Induced Hyperplasia/Dysplasia in the Rag2-/-/Il10-/- Mouse Model (Rag2-/-/Il10-/-小鼠模型中炎症性肠病诱导增生/发育异常的多组学表征)
时间:2020-12-31
期刊:International Journal Of Molecular Sciences
影响因子:IF 6.208
技术平台:RRBS、oxRRBS、RNA-seq、RT-qPCR等
研究摘要:
假设表观遗传失调在炎症性肠病(inflammatory bowel disease,IBD)和结肠癌发展之间的关联中发挥作用。本研究对从肝螺杆菌(H.hepaticus)感染的IBD小鼠模型中收获近端结肠组织,并进行DNA甲基化组、DNA羟甲基化组和转录组分析。RRBS和oxRRBS分析共鉴定出1606个差异甲基化区域(DMR)和3011个差异羟甲基化区域(DhMR),与胃肠疾病、炎性疾病和癌症相关的基因和这些DMR/DhMR重叠。RNA-seq揭示了许多与炎症和癌症相关的基因显著表达变化,包括Duox2、Tgm2、Cdhr5和Hk2在内的几个基因在DNA甲基化/羟甲基化和基因表达水平上均表现出变化。总体而言,本研究结果表明,慢性炎症会引发基因组中DNA甲基化和羟甲基化模式的变化,从而改变关键肿瘤发生基因的表达,并可能导致结直肠癌的发生。
项目设计:
(1)样本选取:
IBD与正常小鼠近侧肠道组织的5mC、5hmC、表达图谱分析
(2)项目设计流程图:
实验结果
(1)肝螺杆菌(H. hepaticus)感染的Rag2-/-/Il10-/-小鼠结肠组织的组织病理学分析
图1:炎症性肠病(IBD)与正常组织的组织病理学检验
- 动物研究设计:Rag2-/-/Il10-/-小鼠在6-7周龄时H.hepaticus感染或无菌培养基处理(对照),并在感染后20周处死
- 感染H.hepaticus(橙色)的Rag2-/-/Il10-/-雄性小鼠和仅用盐水处理的对照小鼠(蓝色)的结肠组织的组织病理学结果:通过对胃肠道不同部位的炎症、水肿、上皮缺陷、隐窝萎缩、增生和发育异常(Dysplasia)的个体评分求和来计算组织学活性指数(n=7)
- 感染或未感染H.hepaticus的Rag2-/-/Il10-/-雄性小鼠胃肠道不同部位的整体5-甲基胞嘧啶(5mC)水平,(盲肠每组n=4,横结肠每组n=3,近端和远端结肠每组n=7)
- 感染或未感染H.hepaticus的Rag2-/-/Il10-/-雄性小鼠胃肠道不同部位的整体5-羟甲基胞嘧啶(5hmC)水平
(2)炎症性肠病(IBD)诱导的增生/发育异常中DNA甲基化和DNA羟甲基化表征
图2:差异甲基化区域(DMR)和差异羟甲基化区域(DhMR)富集的基因在与癌症发生和胃肠疾病相关的基因富集。
- RRBS和oxRRBS技术示意图:通过比较RRBS和oxRRBS数据集分析5mC和5hmC水平。
B-C. 通过RRBS与oxRRBS结合鉴定1606个DMR(B)和3011个DhMR(C)的热图。
D-E. 通过Ingenuity Pathway Analysis(IPA),使用显著性阈值p=0.05的Fisher精确检验,预测受DMR(D)和DhMR(E)基因影响的功能和疾病。
(3)差异甲基化分析揭示IBD诱导的增生/发育异常中存在广泛的DMR和DhMR
图3:Rag2-/-/Il10-/-小鼠在H.hepaticus诱导炎症在单核苷酸分辨率下的甲基化和羟甲基化动态变化
- 784个低DMR,822个高DMR,1454个低DhMR和1557个高DhMR的基因组分布饼图
- DMR/DhMR内每个CpG位点5mC和5hmC的动态变化。0.1是5mC/5hmC水平显著性变化的截止值。数字代表点数量。
表1:H.hepaticus诱导的Rag2−/−/Il10−/−小鼠炎症CpG位点IPA差异甲基化和羟基甲基化的经典通路
(4)IBD诱导增生/发育异常中的基因表达变化
图4:炎症性肠病(IBD)和对照小鼠中近端结肠的转录组分析
- 基于最高方差的500个基因的无监督分层聚类,显示对照组和IBD组之间明显分离
- IPA鉴定上游调控因子Il10ra周围的网络
- 使用显著性阈值p=0.05的Fisher精确检验,预测受差异表达基因影响的疾病和功能
(5)DNA甲基化异常调控差异表达基因的综合分析
图5:IBD诱导的转录组、DNA甲基化组和羟甲基化组变化之间的相关性
A-B. 差异表达且DMR(A)或DhMR(B)富集的基因散点图。颜色表示不同基因组表征,点大小表示每个区域的CpG数量。
- DNA表观遗传标记在IBD诱导的结直肠癌(CRC)发生中的作用的模型。
总结:
本研究采用RRBS+oxRRBS在H.hepaticus感染的IBD Rag2-/-/Il10-/-小鼠模型中,分别绘制了IBD诱导的DNA羟甲基化和DNA甲基化在全基因组中的变化。同时结合RNA-seq基因表达分析,研究结果首次全面揭示了结肠炎症诱导的表观遗传学变化,为进一步研究IBD生物标志物的发现提供了有用的信息,并有可能促进IBD新疗法的未来发展。
关于易基因精准DNA甲基化/羟甲基化测序(oxBS-seq)
羟甲基化5hmC是哺乳动物基因组上的第六碱基,在发育、衰老、神经退行性疾病、复杂疾病及肿瘤发生过程中起重要作用。DNA羟甲基化是近年发现的一种新的DNA修饰并迅速成为研究热点。随着研究的深入,发现之前被认为是检测DNA甲基化标准的重亚硫酸盐测序并不能区分DNA甲基化(5mC)和DNA羟甲基化(5hmC)。
易基因联合剑桥大学建立了化学氧化法结合重亚硫酸盐转化的测序技术(oxidative bisulfite sequencing, oxBS-Seq),该技术不仅可以精确检测DNA甲基化,排除DNA羟甲基化的影响,还可以双文库结合同时单碱基分辨率精确检测DNA羟甲基化。
传统BS转化无法区分5mC和5hmC
传统的Bisulfite测序中,5hmC经过Bisulfite处理后变为CMS,CMS在测序中仍然被读作C碱基,因此不能区分5mC和5hmC。
oxBS技术原理
技术优势:
- DNA甲基化检测全新的“标准”;
- 单碱基检测DNA羟甲基化修饰;
- 多重质控标准检测氧化效率和Bisulfite转换率;
- 实验偏好性低,重复性高(R2>0.98);
- 易基因自主研发的甲基化特异性多重PCR引物设计软件;
- 可满足多种测序应用需求:
- 全基因组氧化甲基化测序(oxWGBS)
- 简化基因组氧化甲基化测序(oxRRBS)
- 目标区域靶基因氧化甲基化测序(Target-oxBS)。
技术路线:
技术指标:
易基因科技提供全面的DNA甲基化研究整体解决方案,技术详情请致电易基因。
参考文献:
Han Q, Kono TJY, Knutson CG, Parry NM, Seiler CL, Fox JG, Tannenbaum SR, Tretyakova NY. Multi-Omics Characterization of Inflammatory Bowel Disease-Induced Hyperplasia/Dysplasia in the Rag2-/-/Il10-/- Mouse Model. Int J Mol Sci. 2020 Dec 31;22(1) pii: ijms22010364.
相关阅读:
一文读懂|精准简化基因组甲基化测序(RRBS+oxRRBS)分析怎么做
功能基因组学的预测网络模型识别炎症性肠病的调节因子
题目英文:A functional genomics predictive network model identifies regulators of inflammatory bowel disease
题目中文:功能基因组学的预测网络模型识别炎症性肠病的调节因子
发表时间:2017年9月11日 杂志:Nature Genetics 影响因子:27.125
摘要英文:A major challenge in inflammatory bowel disease (IBD) is the integration of diverse IBD data sets to construct predictive models of IBD. We present a predictive model of the immune component of IBD that informs causal relationships among loci previously linked to IBD through genome-wide association studies (GWAS) using functional and regulatory annotations that relate to the cells, tissues, and pathophysiology of IBD. Our model consists of individual networks constructed using molecular data generated from intestinal samples isolated from three populations of patients with IBD at different stages of disease. We performed key driver analysis to identify genes predicted to modulate network regulatory states associated with IBD, prioritizing and prospectively validating 12 of the top key drivers experimentally. This validated key driver set not only introduces new regulators of processes central to IBD but also provides the integrated circuits of genetic, molecular, and clinical traits that can be directly queried to interrogate and refine the regulatory framework defining IBD.
摘要中文:炎症性肠病(IBD)的主要挑战在于整合各种IBD数据集以构建IBD预测模型。我们提出了IBD免疫成分的预测模型。该模型通过使用IBD相关的细胞,组织和病理生理学的功能性和调节性注释信息,从而对揭示在先前全基因组关联研究(GWAS)中与IBD相关的基因座之间的因果关系。从三个处于不同疾病阶段的IBD患者队列中获取分子数据,构建出由独立网络组成的模型。我们进行了关键驱动因素分析,以鉴定出那些可以预测与IBD相关的网络调节状态的基因,并对这些基因进行排序以及通过实验对12个最关键的驱动因素进行前瞻性验证。这种经过验证的关键驱动基因集不仅引入了IBD核心过程新的调节器,而且还提供了遗传,分子和临床特征的集成网络,可以直接查询这些特性来询问和完善IBD的调控框架。
背景中文:
克罗恩病(CD)和溃疡性结肠炎(UC)是IBD的主要形式,其特征是肠道炎症复发和缓解。虽然克罗恩病和溃疡性结肠炎的具有不同的临床表型和仅重叠一些分子途径,但它们在很大程度上具有共同的遗传结构。尽管GWAS已鉴定出200多个与IBD相关的基因座,但这些已知的遗传变异仅贡献了约26%的克罗恩病和19%的溃疡性结肠炎的遗传性。
通过构建因果网络模型,可以对大规模、多样的数据进行整合利用,统计推断出任何感兴趣特征之间的因果关系,从而全面描述IBD的结构。已经证明IBD相关基因可以组织形成显著富集免疫和炎症过程的调节网络。
然而,目前为止,还没有从IBD相关组织以及不同IBD疾病阶段的分子状态构建的IBD网络模型。因此,本文整合IBD不同疾病阶段的大规模DNA和RNA数据,以构建IBD病理性炎症成分模型,这有助于区分与IBD相关的炎症成分和肠道的稳态背景功能。
方法中文:(重点描述清楚数据及数据来源)
1)表达数据来自以下三个数据集,并从RNA-seq中鉴定遗传变异。
数据队列 |
样本数量 |
来源 |
备注 |
RISK |
322 |
GSE57945 |
回肠活检样本,包括CD、UC以及control |
CERTIFI |
118 |
GSE100833 |
抗肿瘤坏死因子(TNF)-α治疗无效的CD患者样本,包括多个肠段 |
MSH |
134 |
GSE83687 |
晚期IBD患者样本,多个肠段,包括CD、UC以及control(control取自散发性结肠癌患者正常非炎症肠) |
2)从International Inflammatory Bowel Disease Genetics Consortium (IIBDGC)和Immunochip研究获得CD、UC和IBD基因座(GWAS);整合先前发表的IBD、免疫以及消化相关的eQTL数据集;通过ENCODE、REMC、FANTOM5以及Roadmap的Chip-seq以及DHS-seq等获得不同细胞类型(参与免疫和消化过程)的细胞类型特异性顺式调节元件(CRE)。
3)本文通过获得保守炎症成分(conserved inflammatory component,CIC)的IBD网络,继而进行KDG(key driver gene)分析获得调控子,最后进行实验验证。具体步骤如下:
1、通过整合IBD风险SNP、表达数量性状基因座(eQTL)以及顺式调控元件来识别IBD基因。随后可以在第6步使用这些IBD基因进行KDG的排序。
2、使用RISK、CERTIFI以及MSH的表达谱数据进行加权基因共表达网络分析(WGCNA),从而获得共表达模块。
3、先前研究所描述的巨噬细胞富集的免疫网络(对IBD易感基因以及IBD相关炎症作用)作为基因种子集(seed immune network),以便在IBD组织特异性背景(即不同数据集不同疾病状态下的共表达模块)下识别同源基因集。具体来说,筛选出不同数据集中与免疫网络显著富集的共表达模块(P<0.05),称其为免疫超级模块(immune-super module,亦称tagged module)。
4、通过将每个数据集中的tagged模块进行合并,获得超级模块(super-module)。然后,将不同数据集的超级模块进行相交,得到核心免疫激活模块(core immune activation module,core IAM)。
5、利用三个数据集DNA以及RNA数据构建出各个数据集的贝叶斯网络后,将core IAM或MSS(macrophage-specific signatures)映射到贝叶斯网络获得CIC IBD网络或是MSG(macrophage specific genes)贝叶斯网络。注:巨噬细胞特异性特征(MSS,macrophage specific signatures)是来源于以下三种特征的并集。1)M1,M2和TPP (TNFα, PGE和TPP- (TLR2 ligand P3C))刺激的巨噬细胞共表达模块;2)结核感染的人类DC特征;3)100nM地塞米松治疗的人巨噬细胞特征。此外,使用巨噬细胞特异性基因(MSG,macrophage specific genes)集补充MSS。MSG是在人类巨噬细胞或小鼠小胶质细胞中观察到表达,但在星形胶质细胞、神经元和之前建立的任何M诱导多能干细胞系中均未表达的基因。
6、在CIC IBD网络(或MSG贝叶斯网络)中进行KDG的预测,并利用GWAS IBD基因、VEO(very early onset)IBD基因、免疫网络基因、MSH队列的基因表达特征(CD vs Control, CD vs UC)以及在CERTIFI队列中基因表达与临床特征相关的特征集来进行KDG排序。具体而言,排序可以分为两部分。一是对每个KDG基因映射到CIC IBD网络(或MSG网络),取两个路径长度的基因,获得这些基因与上述那些特征富集的P值,并以P值进行排序;二是对每个KDG基因映射到CIC IBD网络(或MSG网络),取两个路径长度的基因,获得这些基因与上述那些特征显著富集的次数,并以显著富集的次数进行排序。最终,结合两种排序获得综合排序。
7、对KDG基因进行实验验证,使用人类巨噬细胞并对相应KDG-knockdown进行巨噬细胞KDG的验证,以及借助小鼠炎症模型和KDG-knockdown进行CIC IBD的KDG验证。
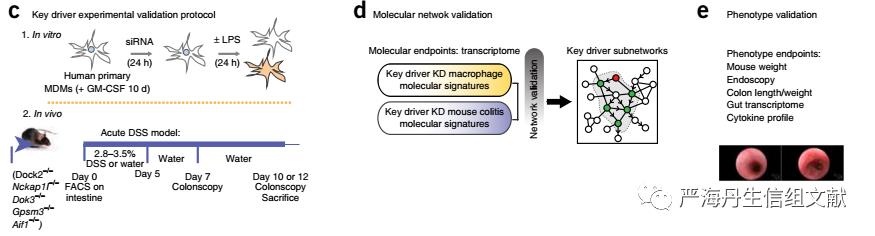
结果中文:
1、对CIC IBD网络或IBD网络中巨噬细胞成分(MSG网络)关键驱动子的识别和排序
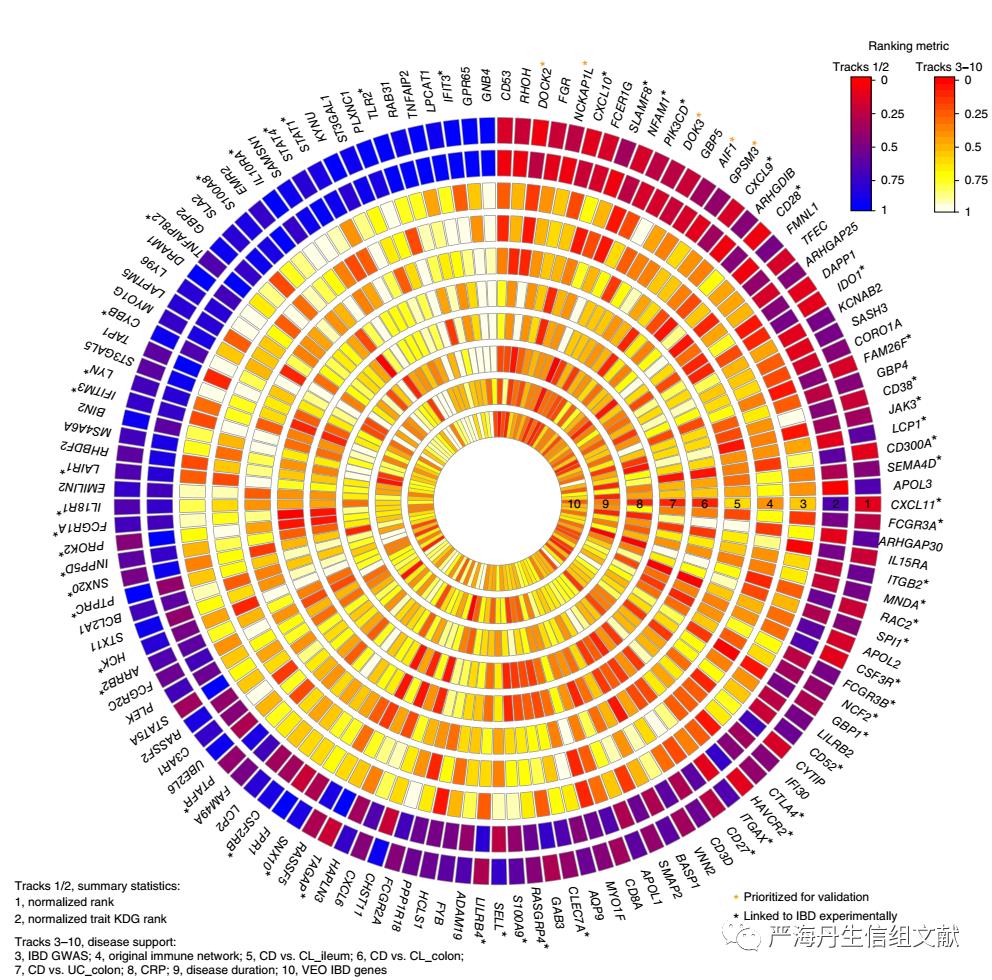
CIC IBD网络中关键驱动子的识别和排序。在将获得的core IMA(228个基因)映射到不同数据集的贝叶斯网络后,获得不同数据集的CIC IBD网络。为了阐明CIC IBD模型的调控框架及其对IBD发病机制,对每个数据集的CIC IBD网络都进行KDG识别,最终共获得133 KDG。在对133个KDG排序后,发现前10%的KDG中有5个KDG(DOCK2, GPSM3, NCKAP1L, DOK3)先前没有被证实与IBD相关。133个KDG排序具体如下图,不同圈表示在不同特征下KDG的排序。
IBD网络中巨噬细胞成分(MSG网络)关键驱动子的识别和排序。鉴于巨噬细胞在肠内稳态中起前哨作用并导致IBD中不适当的炎症反应、seed免疫网络中巨噬细胞富集以及核心IAM中巨噬细胞特异表达的富集,本文更加精确的确定了IBD网络的这一组成部分。
CERTIFI CIC IBD网络是巨噬细胞特征最富集的网络,因此将MSG中的基因投射到CERTIFI IBD网络上,并从这个投影中识别出最大的连接子网络(由这些基因的三个路径长度内的节点组成),获得IBD网络的巨噬细胞特异性成分。对网络进行KDG分析,发现59个KDG(皆在133个CIC IBD网络中识别的KDG),最终选择10个KDG(包括前面5个未被证实与IBD相关的4个基因)进行实验验证。
2、巨噬细胞KDG的体外分子网络验证
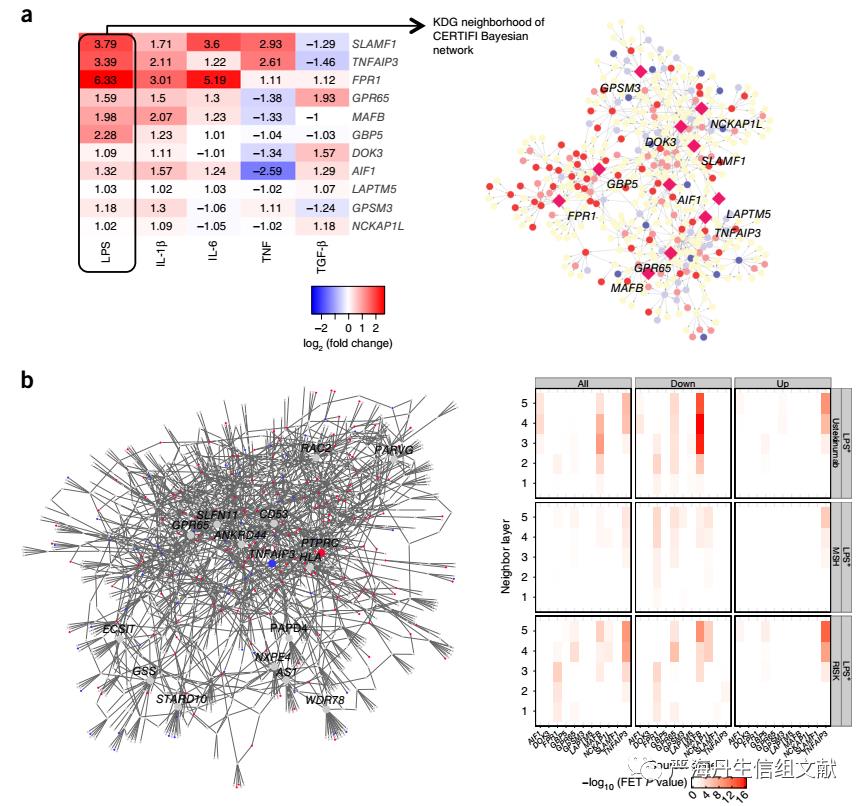
在对候选巨噬细胞KDG进行分子验证时,LPS( lipopolysaccharide,脂多糖)刺激诱导细胞炎症反应时产生最大的差异表达特征并且大多数KDG会对LPS刺激反应(下图a),因此在LPS刺激下,通过比较靶向11个KDG的siRNA与非靶向的siRNA进行KDG的分子验证。发现IBD网络中巨噬细胞特异性成分的相应KDG很好地预测了巨噬细胞特异性KDG-knockdown特征,即巨噬细胞KDG的子网与巨噬细胞特异性KDG-knockdown后表达改变的基因的显著富集(下图b)。
下图a左图表示不同刺激剂与对照组下KDG的倍数改变,a右图表示在LPS刺激下KDG及其邻居节点的倍数改变(菱形表示KDG,颜色表示上下调);下图b左图表示TNFAIP3-knockdown后CERTIFI IBD网络中节点改变的情况,b右图表示在LPS刺激性,相应KDG-knockdown后表达改变的基因与KDG子网富集的情况,纵轴表示取几层作为子网。
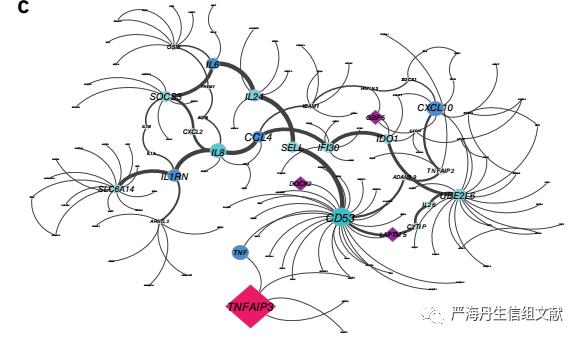
3、巨噬细胞KDG的体外分子网络验证
为了评估KDG在巨噬细胞中扰动的直接功能结果,本文测定了LPS刺激的巨噬细胞上清液中的细胞因子水平(KDG敲除与非靶向对照组相比)。其中GPSM3-knockdown时IL-1RA和CXCL10显著差异表达,NCKAP1L和FPR1-knockdown的IL-6、CCL4和CXCL10的显著差异表达,以及TNFAIP3-knockdown时导致最强的细胞因子差异表达。这些细胞因子(或其受体或配体)与IBD的遗传易感性有关,或被认为是IBD的药物靶点,包括TNF-α、IL-6、IL-1RA47、CXCL10和IL-12Rp40。下图表示TNFAIP3-knockdown后细胞因子的子网,其中红色节点表示TNFAIP3 KDG,紫色节点表示KDG,蓝色节点表示TNFAIP3-knockdown后显著差异表达的细胞因子。
4、肠道KDG的体内分子网络验证
为了了解CIC-IBD网络模型的KDGs是否会影响肠道炎症的易感性,在KDG-knockdown后使用DSS(dextran sulfate sodium)培养小鼠以获得小鼠结肠炎模型从而在体内对KDG进行验证。KDG-knockdown小鼠与野生型对照的差异基因显著富集到IBD相关的分子途径,并且利用表达谱构建的共表达模块也显示出对GWAS IBD基因、VEO IBD基因以及核心IAM的显著富集,表明物种间CIC IBD网络的保守性。此外,本文还进一步测试了给定的KDG的knockdown特征(即KDG-knockdown后表达显著改变的基因)是否会与网络预测的受该KDG控制的一组基因(即KDG特征)显著重叠。除了DOK3外,其他KDG的实验扰动特征会显著地富集IBD网络预测的KDG特征(如下图)。
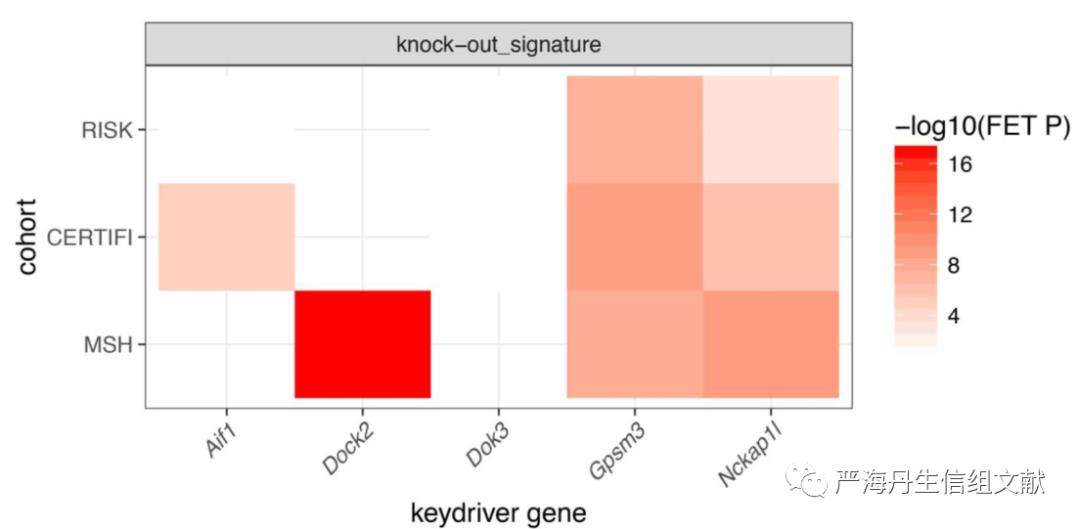
对于每个IBD网络来说,KDG都可以通过KDG特征(即KDG所控制的基因)链接,如下图a中的CERTIFI IBD网络所示。在CERTIFI队列中,与KDG基因表达特征重叠的KDG子网中的基因与那些CRP(C-reactive protein)、乳铁蛋白和钙卫蛋白等临床信息相关的IBD易感基因也显著富集(如下图b,c)。
下图a表示IBD网络中5个KDG的预测转录特征;b图表示GPSM3的子网,其中浅绿色节点表示巨噬细胞IBD CRESNP基因,森林绿色表示DSS培养下GPSM3-knockdown的特征,亮绿色表示这两者兼之,蓝色边框节点表示CRP(C-reactive protein)、乳铁蛋白和钙卫蛋白等临床信息相关的基因,红色节点表示ustekinumab的分子靶标;c图表示DOCK2的子网,其中浅绿色节点表示T细胞IBD CRESNP基因,森林绿色表示DOCK2-knockdown的上调特征,亮绿色表示这两者兼之,蓝色边框节点表示CRP(C-reactive protein)、乳铁蛋白和钙卫蛋白等临床信息相关的基因,红色节点表示与ustekinumab相关。
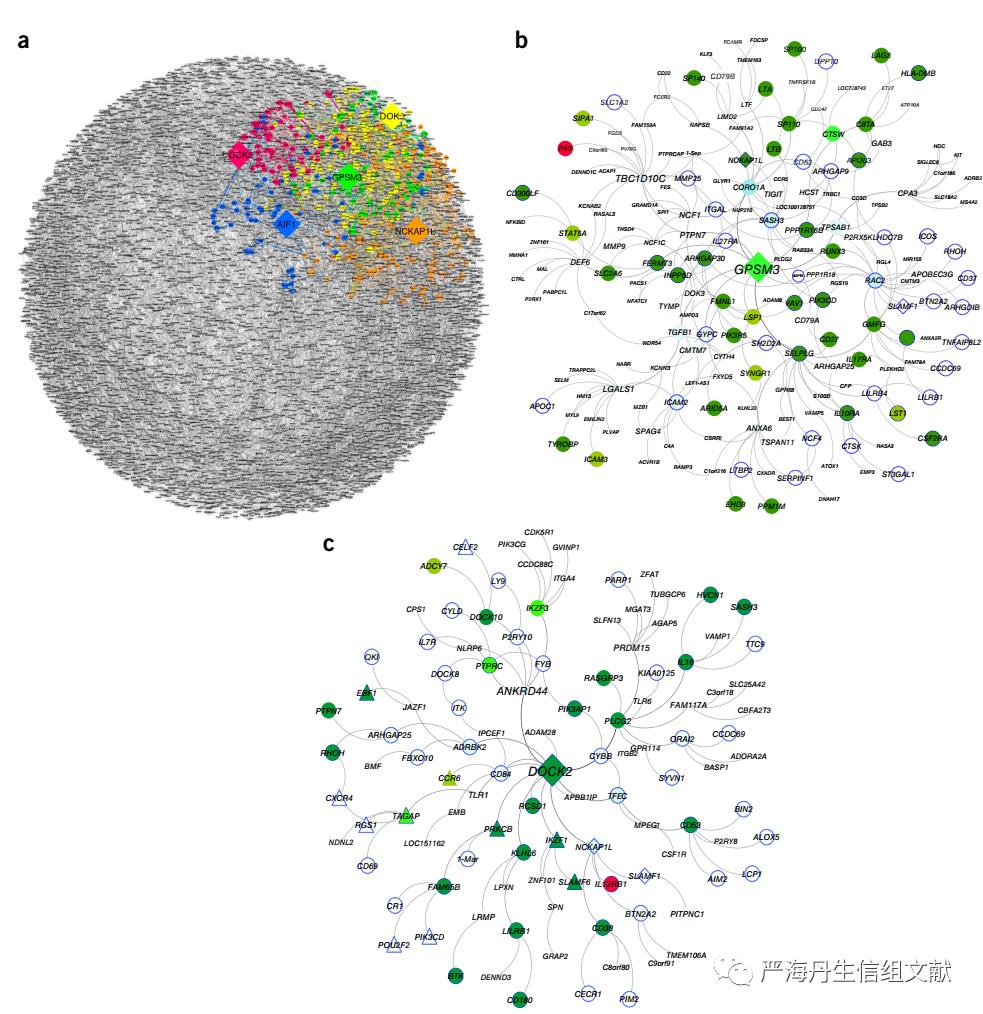
5、肠道KDG的体内验证
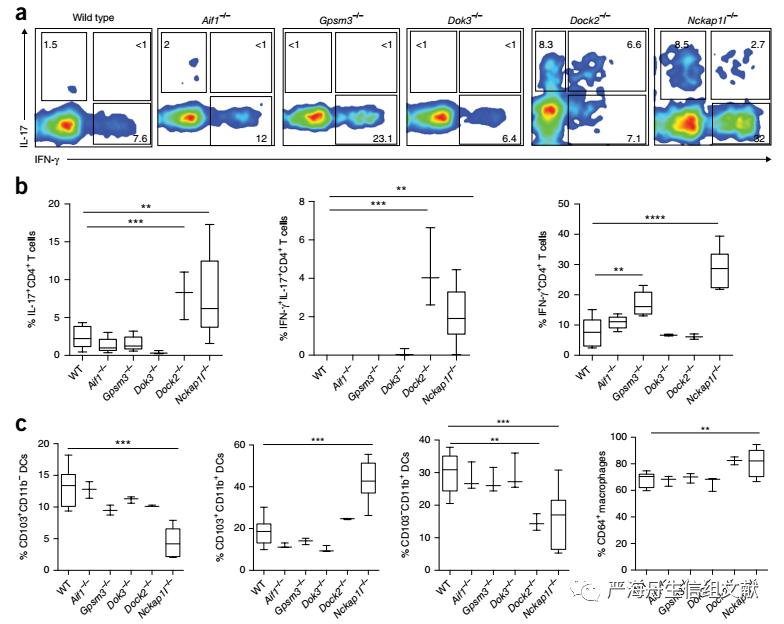
为了验证肠道KDGs与IBD的相关性,我们首先研究了扰动这些基因是否会破坏免疫稳态。对于T细胞,本文评估了CD4+T细胞中IFN-γ和IL-17A的产生(两者都与克罗恩病的病理学有关)。与野生型同窝对照组相比,Nckap1l−/−小鼠、Dock2−/−小鼠和Gpsm3−/−小鼠肠道固有层中IL-17A+和/或IFN-γ+CD4+T细胞的频率存在显著差异(下图a, b)。对于髓系细胞,使用抗CD103、CD11b和CD64的抗体定义了肠道树突状细胞(DC)和巨噬细胞的功能亚群。Nckap1l−/−小鼠的CD103+CD11b+细胞频率升高,而Nckap1l−/−和Dock2−/−小鼠的CD11b+单阳性DC频率降低,同时Nckap1l−/−小鼠的CD103+DC频率降低,CD64+巨噬细胞频率升高,Dok3−/−,Gpsm3−/−,和Aif1−/−小鼠在结肠中的这些髓细胞亚群的免疫细胞比率没有显著差异(下图c)。
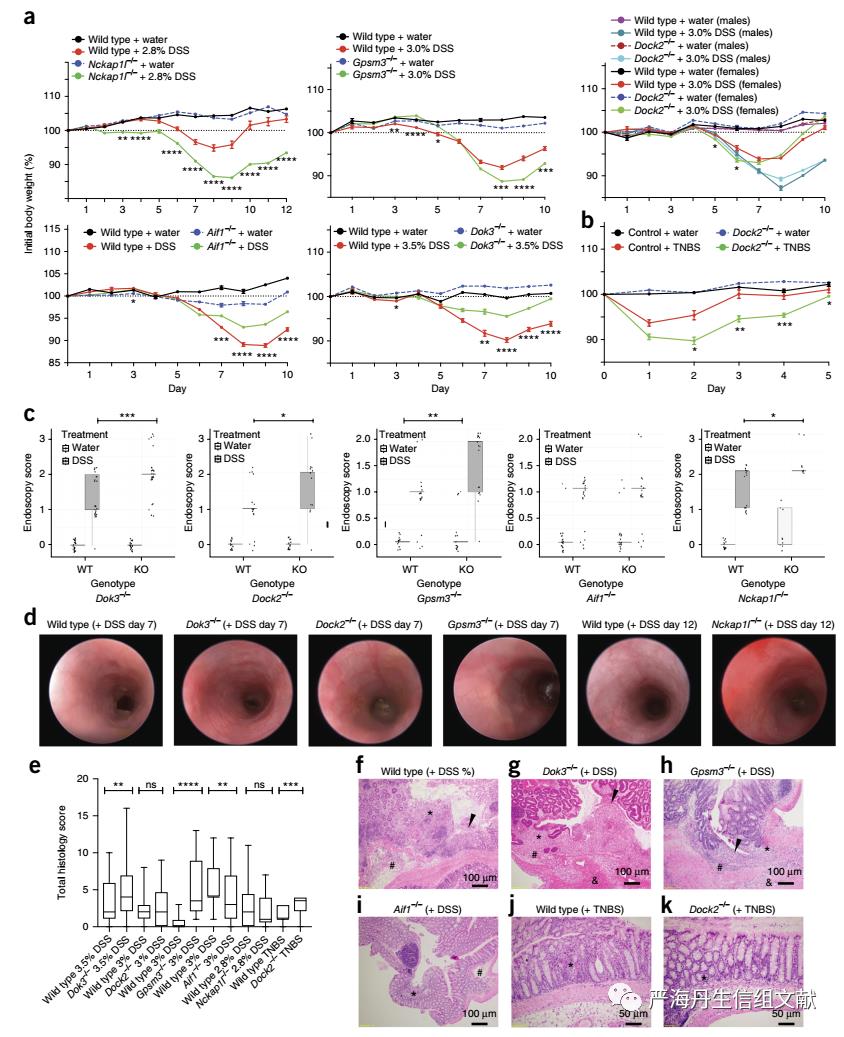
为了确定KDGs是否影响肠道炎症的易感性,本文采用了DSS、TNBS等小鼠结肠炎模型。野生型同窝对照小鼠在DSS、TNBS培养后出现肠道炎症症状。与同窝鼠对照组相比,Nckap1l−/−和Gpsm3−/−小鼠在DSS治疗后有更严重的体重减轻和更高的内镜评分(下图a, b)。在DSS治疗下,Aif1−/−和Dok3−/−缓解小鼠的体重减轻(下图a)。与野生型同窝对照相比,DSS治疗下Dock2-/-,Gpsm3-/-,Dok3-/-和Nckap1-/-小鼠均表现出显著更差的内窥镜评分(下图c,d)。 同时与野生型对照相比,Dok3-/-和Gpsm3-/-小鼠表现出显著更差的组织学评分(下图e),而Aif1-/-小鼠表现出显着降低的病理学评分(下图f-k)。
总体来说,本文检测的每只KDG-knockdown小鼠在结肠炎中至少表现出显著的体重减轻表型和肠道炎症表型在内的一种表型(如下表)。
讨论中文:
有人支持KDG在疾病中的机械作用是由网络的调节状态改变所驱动的,而这调节状态的改变受到免疫有关过程的明确影响。本文确定的KDG中存在一个统一主题,即RAC激活和细胞骨架重排,而这是免疫过程中的一个中心介质,与克罗恩病、实验性结肠炎以及溃疡性结肠炎有关(如下图NF-κB途径,RAC及其肌动蛋白细胞骨架重排,RAS,NLRP3炎性小体途径以及TLR和趋化因子受体均由本文确定的KDG调节)。本文通过构建预测网络模型识别KDG以及使用生物实验验证了KDG的合理敏感性,但是对于相应的特异性理解是一项十分困难的任务。
以上是关于易基因: oxRRBS+RRBS揭示炎症性肠病导致发育异常的表观遗传机制|甲基化研究的主要内容,如果未能解决你的问题,请参考以下文章
易基因:MeRIP-seq等揭示m6A reader YTHDF1在结直肠癌PD-1免疫治疗中的作用|Gut
易基因:全基因组ChIP-seq分析揭示细菌转录因子PhoB的基因内结合位点|mBio
易基因:MeRIP-seq等揭示m6A甲基化修饰对抗病毒基因表达的转录调控机制|Cell Rep
易基因:DNA甲基化和转录组分析揭示野生草莓干旱胁迫分子调控机制|植物抗逆