Nature:哈佛&牛津开发基因致病性预测模型,现已成功预测3600万个致命基因突变...
Posted QbitAl
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了Nature:哈佛&牛津开发基因致病性预测模型,现已成功预测3600万个致命基因突变...相关的知识,希望对你有一定的参考价值。
博雯 发自 凹非寺
量子位 报道 | 公众号 QbitAI
直接从基因层面预测疾病,这一直是近现代医学研究的主要方向之一。
然而,全体人类的基因变异体数量远超现有的探测技术,甚至仅仅是不同个体的蛋白质区编码也会展现出巨大的差异性。
因此,超过98%的基因变异给人体带来的影响依旧是未知且无法预测的。
但最近,来自哈佛医学院和牛津大学的科学家合作开发了一种AI模型,成功预测了3219个疾病基因中超过3600万个变体的致病性,并将超过25万个未知变体进行了归类。

这项研究现已登上Nature。
“从进化中预测致病性”
其实,现在临床上已有用于预测基因变异影响的模型。
但这些模型往往是在经过标注的临床数据集上进行有监督学习,一旦进入现实场景,标签偏差、标签稀疏以及噪音就会造成其准确率的下降,并不能作为基因变异体分类的可靠依据。
而这次的研究团队提出了一个叫做EVE(Evolutionary model of Variant Effect)的模型。
这是一个仅根据进化序列训练的无监督生成模型。
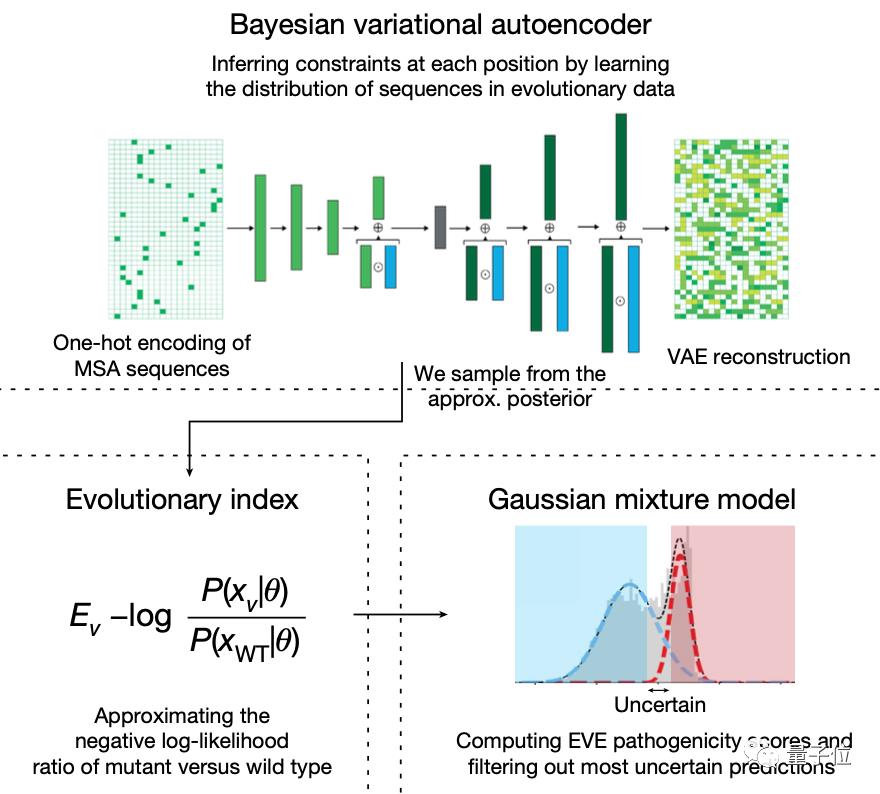
模型预测变异基因的致病性主要分为两步:
第一步,使用变型自动编码器VAE来学习蛋白质的氨基酸序列分布。
学习了多个领域的复杂高维分布之后,模型就捕捉到了进化过程中的自然序列约束,包括各种位置之间的复杂依赖关系。
再从得到的近似后验分布(Approximate Posterior Distribution)中取样,评估每个单一氨基酸变体相对于野生型的相对可能性。
这种相对可能性被称为“进化指数”,与临床标签进行比较后发现,区分致病性和良性标签的数值在不同的蛋白质中是一致的,这说明无监督的方法能够有效推断致病性。
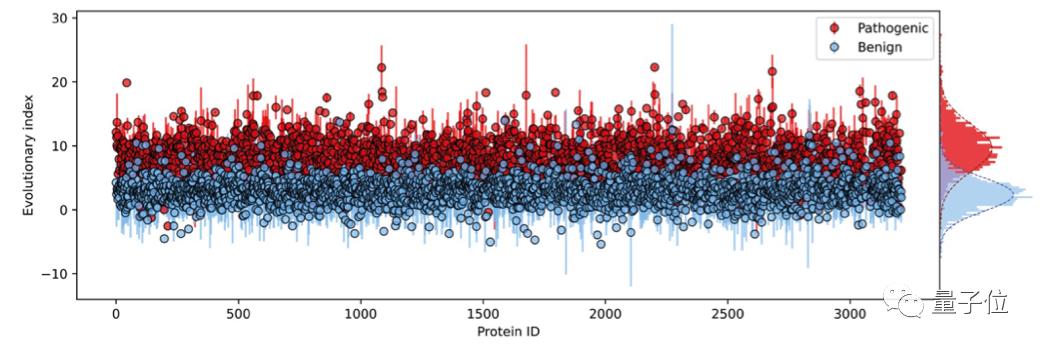
第二步,在所有单一氨基酸变体的进化指数分布上拟合了一个双组分(two-component)的全局-局部高斯混合模型。
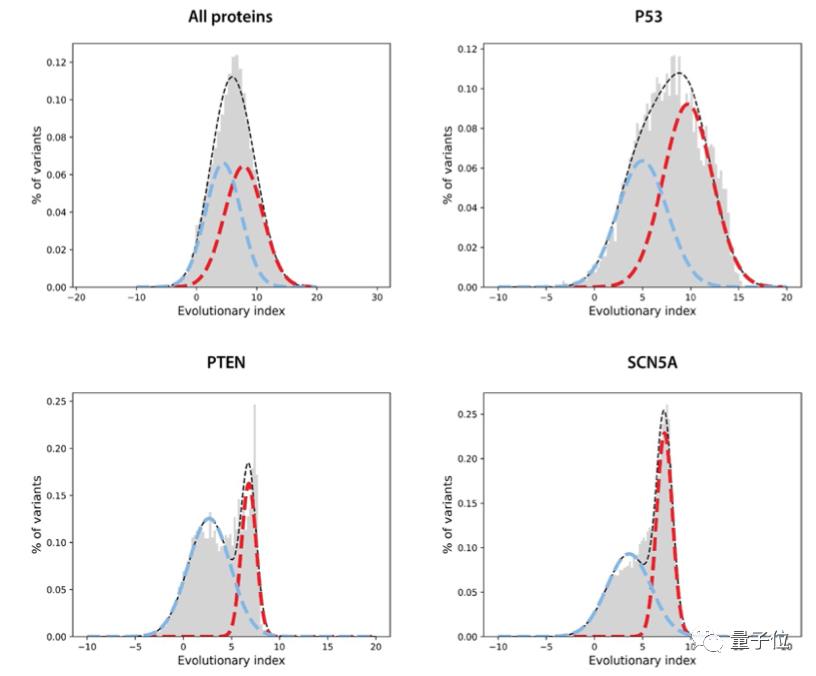
这一步的输出是在区间[0,1]内定义的连续致病性值,0代表良性,1代表致病性。
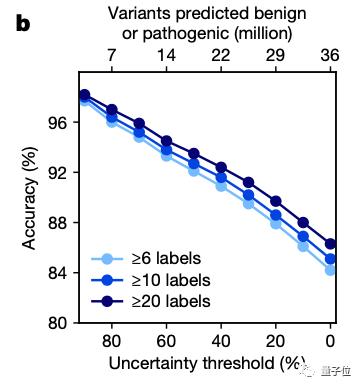
然后将EVE模型运用于ClinVar数据库中的3219个人类基因上,得到的结果图中的平均曲线面积(AUC)为0.91,说明EVE模型对绝大多数的基因变异都能做到具有临床意义的预测:
优于已知模型,与实验预测效果一致
研究团队也将EVE模型与已知的模型进行了对比,可以看到,在预先确定已知的已标注临床数据的预测上,其效果优于同类计算模型:
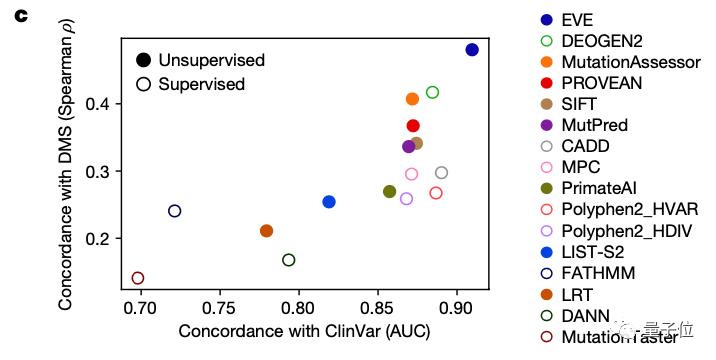
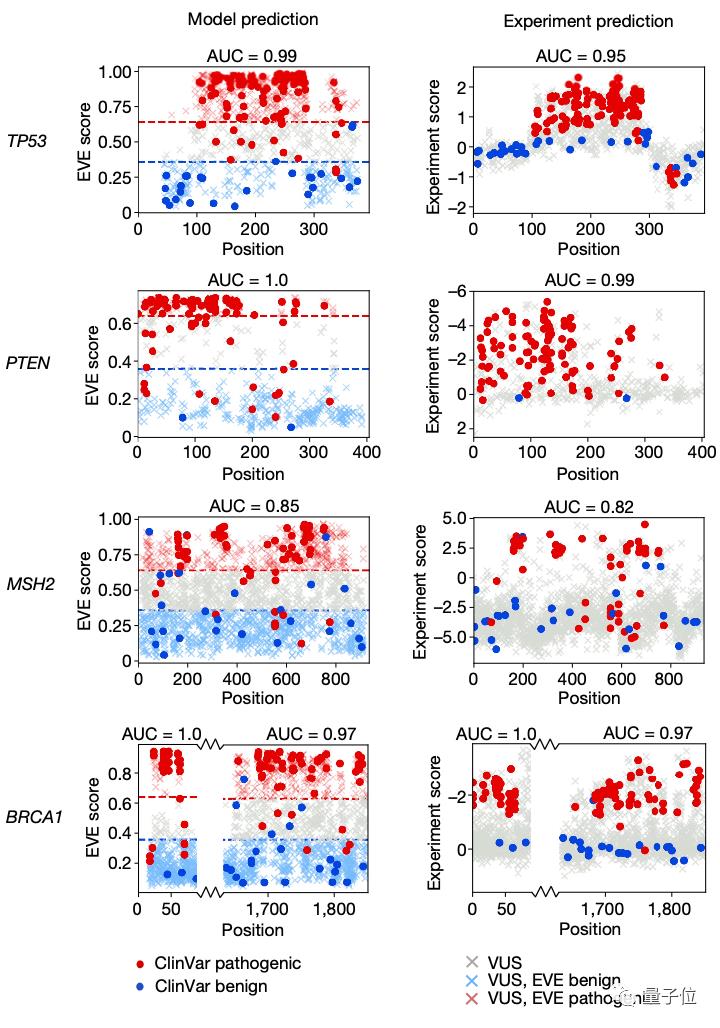
那么这样一个AI计算模型与用于预测致病性的经典方法——深度突变扫描实验(Deep Mutational Scan Experiment)相比效果又如何呢?
对比实验后可以看到,EVE模型在临床预测方面的总体表现与经典方法效果基本一致:
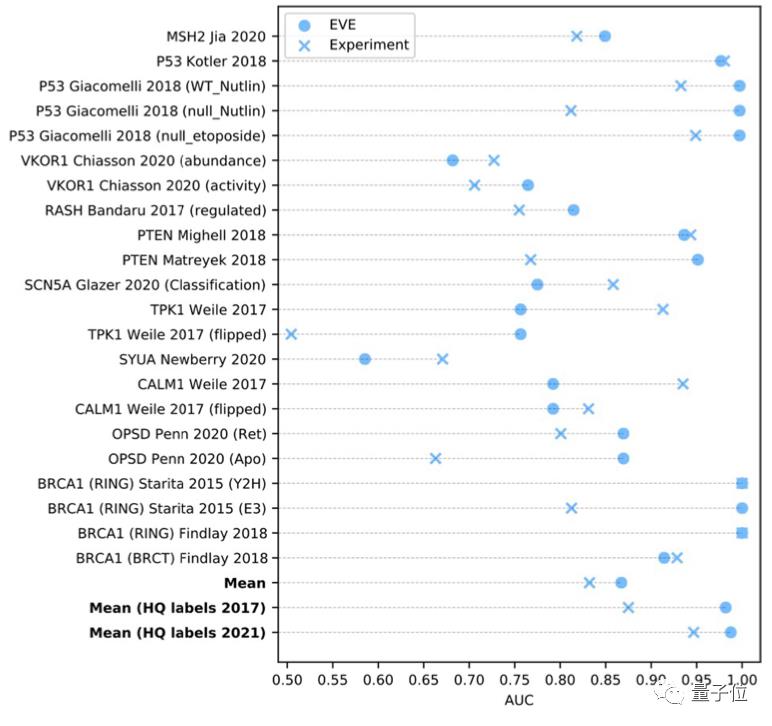
而当从ClinVar数据库中选择一组数量规模更大,但高质量标注较小的数据时,EVE模型的表现甚至更好:
哈佛&牛津合作出品
这篇论文有三位共同一作,其中Jonathan Frazer和Mafalda Dias都来自哈佛大学的系统生物学,他们同时也是Marks Group实验室中的一员。


而Pascal Notin则是来自牛津大学的计算机科学专业的博士生,主要研究领域包括贝叶斯深度学习、生成模型、因果推理和计算生物学的交叉领域。

论文链接:
https://www.nature.com/articles/s41586-021-04043-8
以上是关于Nature:哈佛&牛津开发基因致病性预测模型,现已成功预测3600万个致命基因突变...的主要内容,如果未能解决你的问题,请参考以下文章
类脑芯片怎么搞?三星&哈佛:直接复制粘贴神经元 | Nature子刊
针灸研究登上Nature:哈佛团队找出刺激穴位治疗疾病背后原理
自然语言处理加速ACMG解读:以顾卫红老师的帖子“PLA2G6基因的2个变异”为例