GWAS基本分析内容
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GWAS基本分析内容相关的知识,希望对你有一定的参考价值。
参考技术A 之前给大家大致介绍了GWAS在临床生信分析中的概况,包括一些基本概念,原理和注意事项(出门左手边—> 临床生物信息学中的GWAS分析 ),这次具体讲讲 GWAS基本分析内容及结果解读 (这篇也是我在百迈客云课堂的个人学习笔记,有兴趣学习的可以去购买课程 百迈客GWAS生物信息培训课程 )另外参考文献及配图来自
GWAS基本分析内容
1)按分型百分比过滤
一般剔除缺失率在20%以上的位点,如果数据量比较大可以放宽到50%
2)按等位基因频率过滤
去除第二等位基因频率小于5%的位点,如果数据量比较大可以放宽到1%
3)多等位位点的过滤
根据软件的需要,有些软件不支持多等位位点
4)哈迪温伯格平衡过滤
人类case/control中一般将不符合哈迪温伯格平衡的位点过滤掉,动植物不使用该过滤
5)极端表型的去除
最低饱和标记量=基因组大小/LD衰减距离
密度越大越好:检测到功能位点的概率增大;处于同一block的位点相互验证
可以根据LD衰减距离来决定候选基因上下游的范围
1)目的:对群体结构和亲缘关系进行评估以确定使用的统计模型和获得相应的矩阵
2)评估内容(遗传上差异过大应剔除,相似性高的保留其一)
群体结构: 构建系统发育树 (必备)
同一物种内序列差异不大构建NJ树(mega),序列差异较大,不同种构建ML树(RAxML),贝叶斯树(ExaBayes)
model-base的群体结构分析
主成分分析
亲缘关系:相似性系数热图、遗传距离
3)群体结构和亲缘关系是导致关联结果出现 假阳性 的两个主要因素(课程里谢坤大牛用实例说明了群体结构和亲缘关系的选择对结果的巨大影响,比如玉米Dwarf8基因 Lessons from Dwarf8 on the Strengths and Weaknesses of Structured Association Mapping )
1)选择正确的统计方法:
小标记量:简单的t-test或ANOVA
case/control质量性状:卡方检验, OR检验,逻辑回归
数量性状:一般使用多种模型(GLM/MLM/EMMAX/fast-LMM)同时分析
2)确定显著性阈值
Bonferroni correction
3)结果解读
可视化:曼哈顿图
分位点图(随机预测出的P和实际算出来P的比较,是否异常显著)
4)若结果不显著:
可能原因及解决办法 :
性状考察不准确——提供准确表型
性状受环境影响大——多年多点重复
性状由多个小效应位点控制——增加样本量
模型检测效力power不够——更换合适的模型
标记密度不够——更加标记量
实在不行就忽略阈值,选择有明显的峰值区域进行验证
获得显著位点后:
1)进行LD block分析,确定候选区间的范围
2)对候选区间内的基因做功能注释(nr,GO,KEGG等)
3)显著位点是否位于编码区,是否是非同义突变
4)同源分析,结合其他物种对应的同源基因的功能来猜测候选基因的功能
RT-PCR、蛋白表达、转基因功能验证、RNA干扰
高分文章实例: OsSPL13 controls grain size in cultivated rice
绘图之全基因组关联分析可视化(GWAS)
点击关注,桓峰基因
桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
134篇原创内容
公众号
桓峰基因公众号推出基于基因组数据生信分析教程并配有视频在线教程,目前整理出来的教程目录如下:
DNA 1. Germline Mutation Vs. Somatic Mutation 傻傻分不清楚
DNA 2. SCI 文章中基因组变异分析神器之 maftools
DNA 3. SCI 文章中基因组变异分析神器之 maftools
DNA 4. SCI 文章中基因组的突变信号(maftools)
DNA 5. 基因组变异文件VCF格式详解
DNA 6. 基因组变异之绘制精美瀑布图(ComplexHeatmap)
DNA 7. 基因组拷贝数变异分析及可视化 (GISTIC2.0)
DNA 8. 癌症的突变异质性及寻找新的癌症驱动基因(MutSigCV)
DNA 9. 揭秘肿瘤异质性与TMB, MSI之间的相关性
DNA 10. 识别癌症驱动基因 (OncodriveCLUST)
DNA 11. 识别肿瘤蛋白质三维结构上突变热点(HotSpot3D)
DNA 12. SCI 文章绘图之全基因组关联分析可视化(GWAS)
这期介绍一下GWAS分析的可视化软件包CMplot,对于研究SNP全体分析,家系遗传等方向的非常实用,给自己的文章添点色彩感。

我们来看看这个软件包能做的事情,GWAS可视化算是很OK了,记得这方面的分析可以考虑使用了!
前言
全基因组关联分析(GWAS)是在某一特定人群中研究遗传突变和表型之间的相关性。GWAS的理论基础是连锁不平衡定律(linkage disequilibrium,LD),既假设观察到的SNP与真正的致病突变(causal variant)之间存在很强的LD全基因组关联分析(Genome wide association study,GWAS)是对多个个体在全基因组范围的遗传变异(标记)多态性进行检测,获得基因型,进而将基因型与可观测的性状,即表型,进行群体水平的统计学分析,根据统计量或显著性 p 值筛选出最有可能影响该性状的遗传变异(标记),挖掘与性状变异相关的基因。因此,GWAS分析需要准备的文件主要有:基因型数据和表型数据。
表型数据统计分析方法包括:
-
逻辑回归(表型数据为二元)
-
线性回归(表型数据为连续性变量)
-
表型数据正态分析(如果不是正态分布,需转换处理为正态分布)
-
表型数据均值、中值、最大值、最小值
-
影响因子对表型的影响分析
基因型数据一般是测序下机数据经SNP calling、基因型填补等后拿到的.vcf文件,后续根据研究者的要求和条件进行质控(Quality control),群体结构分层及亲缘关系/PCA等处理。
软件安装
我们使用CMplot软件包来实现GWAS绘制Manhattan图
if (!require(CMplot)) install.packages("CMplot")
library("CMplot")
参数说明
这个CMplot函数的参数还是蛮多的,毕竟是绘制这种复杂的图像,这也是避免不了的事情,我们大概解释一下参数。
Usage CMplot(Pmap, col=c(“#4197d8”, “#f8c120”, “#413496”, “#495226”, “#d60b6f”, “#e66519”, “#d581b7”, “#83d3ad”, “#7c162c”, “#26755d”), bin.size=1e6, bin.range=NULL, bin.legend.num=10, pch=19, type=“p”, band=1, H=1.5, ylim=NULL, cex.axis=1, lwd.axis=1.5, cex.lab=1.5, plot.type=“b”, multracks=FALSE, points.alpha=100L, cex=c(0.5,1,1), r=0.3, outward=FALSE, ylab=expression(-log[10](italic§)), ylab.pos=3, xticks.pos=1, mar = c(3,6,3,3), threshold = NULL, threshold.col=“red”, threshold.lwd=1, threshold.lty=2, amplify= TRUE, signal.cex = 1.5, signal.pch = 19, signal.col=NULL, signal.line=2, highlight=NULL, highlight.cex=1, highlight.pch=19, highlight.type=“p”, highlight.col=“red”, highlight.text=NULL, highlight.text.col=“black”, highlight.text.cex=1, highlight.text.xadj=NULL, highlight.text.yadj=NULL, highlight.text.font=3, chr.labels=NULL, chr.border=FALSE, chr.labels.angle=0, chr.den.col=“black”, chr.pos.max=FALSE, cir.band=1, cir.chr=TRUE, cir.chr.h=1.5, cir.legend=TRUE, cir.legend.cex=0.6, cir.legend.col=“black”, LOG10=TRUE, box=FALSE, conf.int=TRUE, conf.int.col=NULL, file.output=TRUE, file=c(“jpg”,“pdf”,“tiff”), dpi=300, height=NULL, width=NULL, memo=“”, main=“”, main.cex=1.5, main.font=2, trait.legend.ncol=NULL, verbose=TRUE)
参数细节如下:
Pmap 输入数据文件
col 设置颜色,可以是vector or matrix
当vector数量少于染色体数目时,循环利用;也可以对每一个性状的每一条染色体进行设置
bin.size 设置SNP密度图中的窗口大小
bin.max bin中SNP数量的阈值,当大于阈值时染色体bin颜色为同一颜色
cex 设置绘制点的大小
pch 设置绘制点的形状,同plot中的"pch"
band 设置染色体之间的间隔,当为0时染色体间无空隙,默认为1
cir.band 设置不同circle的空隙,默认为1
H 性状circle的高度设置,默认1
ylim 设置y轴的范围同plot中的"ylim"
cex.axis 设置坐标轴字体和标签字体的大小
bin.size 设置SNP密度图中的窗口大小
cex.axis 设置坐标轴字体和标签字体的大小
plot.type 设置不同的绘图类型,可以设定为 “d”, “c”, “m”, “q” or “b”
“d” 表示 SNP density plot “c” 表示 circle-Manhattan plot
“m” 表示 Manhattan plot “q” 表示 Q-Q plot
“b” 表示 circle-Manhattan, Manhattan and Q-Q plots一起绘制
plot.type=c(“m”,“q”) 表示Manhattan plot和Q-Q plot一起绘制
multracks 设置是否需要绘制多个track
cex 绘制点的大小,可是为单个数值或向量(对应同一绘图中不同的plot)
r 设置圈的半径大小
xlab 设置x轴标签
ylab 设置y轴标签
outward 设置点的朝向是否向外
threshold 设置阈值并添加阈值线
threshold.col 设置阈值线的颜色
threshold.lwd 设置阈值线的宽度
threshold.lty 设置阈值线的类型
amplify 设置是否放大显著的点
chr.labels 设置染色体的标签
signal.cex 设置显著点的大小
signal.pch 设置显著点的形状
signal.col 设置显著点的颜色
singal.line 设置显著线的宽度
chr.den.col 设置SNP密度图的颜色
cir.band 设置环状曼哈顿图中不同染色体之间的间隔
cir.chr 设置是否显示染色体的边界
cir.chr.h 设置染色体边界的高度
chr.den.col SNP密度的颜色,可以为字符,向量或空
cir.legend 设置是否显示图例
cir.legend.cex 设置图例字体的大小
cir.legend.col 设置图例的颜色
LOG10 设置是否对p-value取log10对数
box 曼哈顿是否绘制方框
conf.int.col 设置QQ图中置信区间的颜色
file.output 设置是否输出图片
file 设置输出图片的格式,可以设定为"jpg", “pdf”, “tiff”
dpi 设置输出图片的分辨度
memo 设置输出图片文件的名字
数据读取
我们这里选择使用软件包自带的两个数据集,方便大家理解。
Genotyped by pig 60k chip Description This dataset gives the results of Genome-wide association study of 3 traits, individuals were genotyped by pig 60K chip. Format A dataframe containing 3 traits’ Pvalue
data(pig60K) #calculated p-values by MLM
head(pig60K)
## SNP Chromosome Position trait1 trait2 trait3
## 1 ALGA0000009 1 52297 0.7738187 0.51194318 0.51194318
## 2 ALGA0000014 1 79763 0.7738187 0.51194318 0.51194318
## 3 ALGA0000021 1 209568 0.7583016 0.98405289 0.98405289
## 4 ALGA0000022 1 292758 0.7200305 0.48887140 0.48887140
## 5 ALGA0000046 1 747831 0.9736840 0.22096836 0.22096836
## 6 ALGA0000047 1 761957 0.9174565 0.05753712 0.05753712
Genotyped by Bovine50K chip Description This dataset gives the effects of all SNPs using rrBLUP algorithm for 3 traits, individuals were genotyped by Bovine50K chip. Format A dataframe containing 3 traits’ SNPs effects
data(cattle50K) #calculated SNP effects by rrblup
head(cattle50K)
## SNP chr pos Somatic cell score Milk yield Fat percentage
## 1 SNP1 1 59082 0.000244361 0.000484255 0.001379210
## 2 SNP2 1 118164 0.000532272 0.000039800 0.000598951
## 3 SNP3 1 177246 0.001633058 0.000311645 0.000279427
## 4 SNP4 1 236328 0.001412865 0.000909370 0.001040161
## 5 SNP5 1 295410 0.000090700 0.002202973 0.000351394
## 6 SNP6 1 354493 0.000110681 0.000342628 0.000105792
实例操作
这里的例子都是来之文档说明,我就当一次搬运工,有需要使用自己数据分析的,搞不定可以找桓峰基因,我们随时在线等!话不多说,上代码就可以了。
使用例子:https://github.com/YinLiLin/CMplot
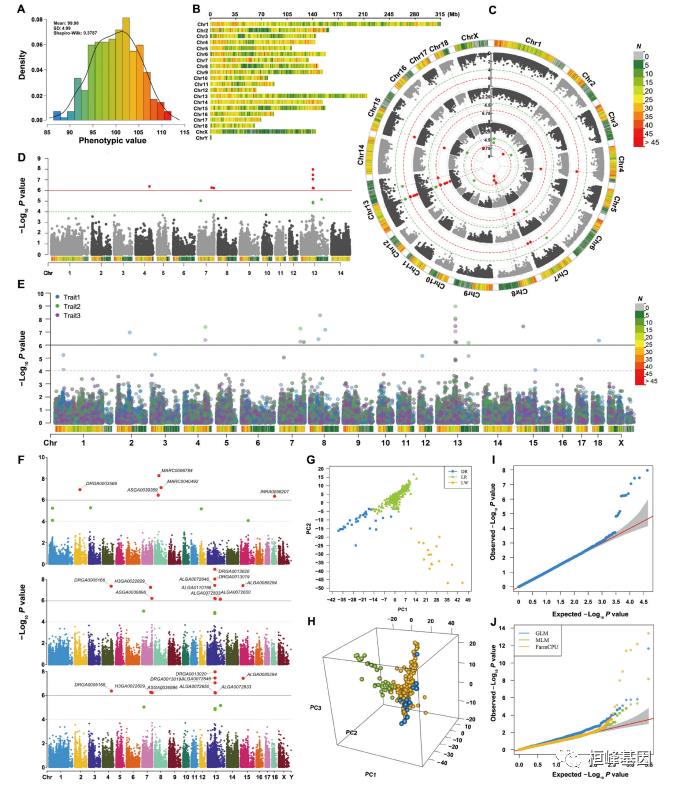
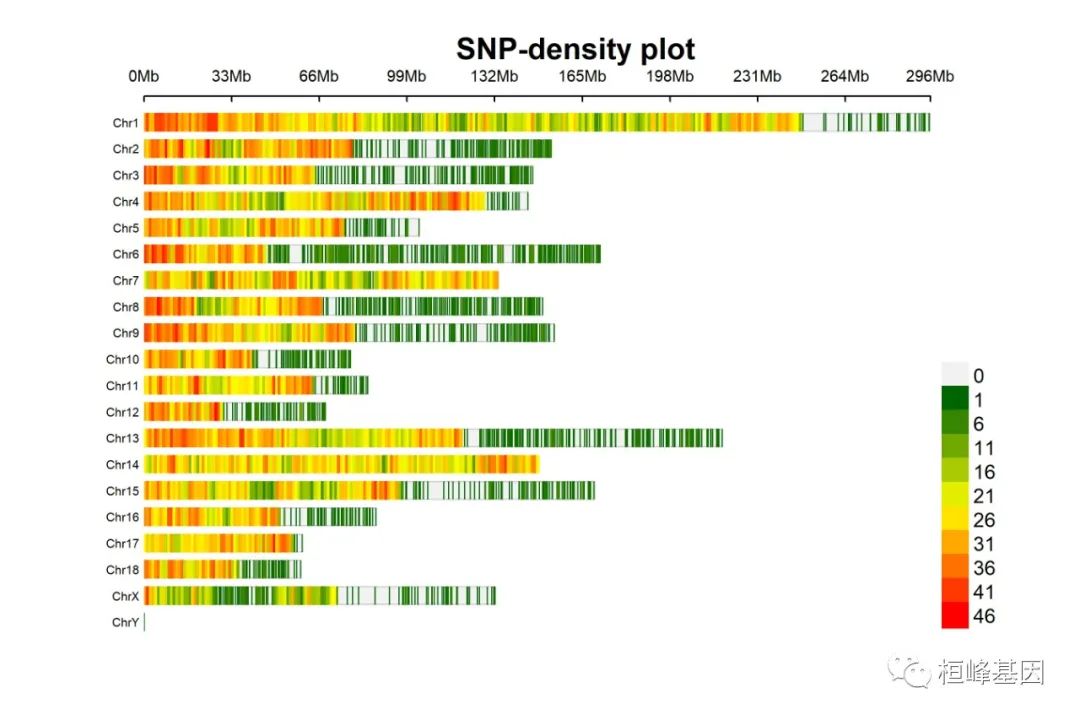
1. SNP-density plot
绘制SNP密度图
CMplot(pig60K, type = "p", plot.type = "d", bin.size = 1e+06, chr.den.col = c("darkgreen",
"yellow", "red"), file = "jpg", memo = "", dpi = 300, main = "SNP-density plot",
file.output = TRUE, verbose = TRUE, width = 9, height = 6)
## SNP-Density Plotting.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot
2. Circular-Manhattan plot
绘制圆形Manhattan图
(1) Genome-wide association study(GWAS)
CMplot(pig60K, type = "p", plot.type = "c", chr.labels = paste("Chr", c(1:18, "X",
"Y"), sep = ""), r = 0.4, cir.legend = TRUE, outward = FALSE, cir.legend.col = "black",
cir.chr.h = 1.3, chr.den.col = "black", file = "jpg", memo = "", dpi = 300, file.output = TRUE,
verbose = TRUE, width = 10, height = 10)
## Circular-Manhattan Plotting trait1.
## Circular-Manhattan Plotting trait2.
## Circular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot

添加色带的Manhattan图
CMplot(pig60K, type = "p", plot.type = "c", r = 0.4, col = c("grey30", "grey60"),
chr.labels = paste("Chr", c(1:18, "X", "Y"), sep = ""), threshold = c(1e-06,
1e-04), cir.chr.h = 1.5, amplify = TRUE, threshold.lty = c(1, 2), threshold.col = c("red",
"blue"), signal.line = 1, signal.col = c("red", "green"), chr.den.col = c("darkgreen",
"yellow", "red"), bin.size = 1e+06, outward = FALSE, file = "jpg", memo = "",
dpi = 300, file.output = TRUE, verbose = TRUE, width = 10, height = 10)
## Circular-Manhattan Plotting trait1.
## Circular-Manhattan Plotting trait2.
## Circular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot

(2) Genomic Selection/Prediction(GS/GP)
基因组选择或者预测
CMplot(cattle50K, type = "p", plot.type = "c", LOG10 = FALSE, outward = TRUE, col = matrix(c("#4DAF4A",
NA, NA, "dodgerblue4", "deepskyblue", NA, "dodgerblue1", "olivedrab3", "darkgoldenrod1"),
nrow = 3, byrow = TRUE), chr.labels = paste("Chr", c(1:29), sep = ""), threshold = NULL,
r = 1.2, cir.chr.h = 1.5, cir.legend.cex = 0.5, cir.band = 1, file = "jpg", memo = "",
dpi = 300, chr.den.col = "black", file.output = TRUE, verbose = TRUE, width = 10,
height = 10)
## Circular-Manhattan Plotting Somatic cell score.
## Circular-Manhattan Plotting Milk yield.
## Circular-Manhattan Plotting Fat percentage.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot

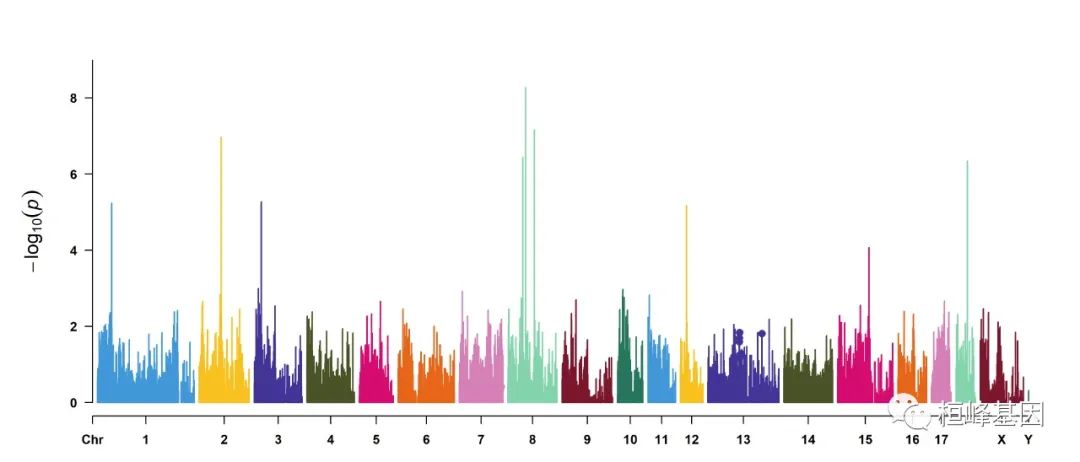
3. Single_track Rectangular-Manhattan plot
1. Genome-wide association study(GWAS)
CMplot(pig60K, type = "p", plot.type = "m", LOG10 = TRUE, threshold = NULL, file = "jpg",
memo = "", dpi = 300, file.output = TRUE, verbose = TRUE, width = 14, height = 6,
chr.labels.angle = 45)
## Rectangular-Manhattan Plotting trait1.
## Rectangular-Manhattan Plotting trait2.
## Rectangular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot


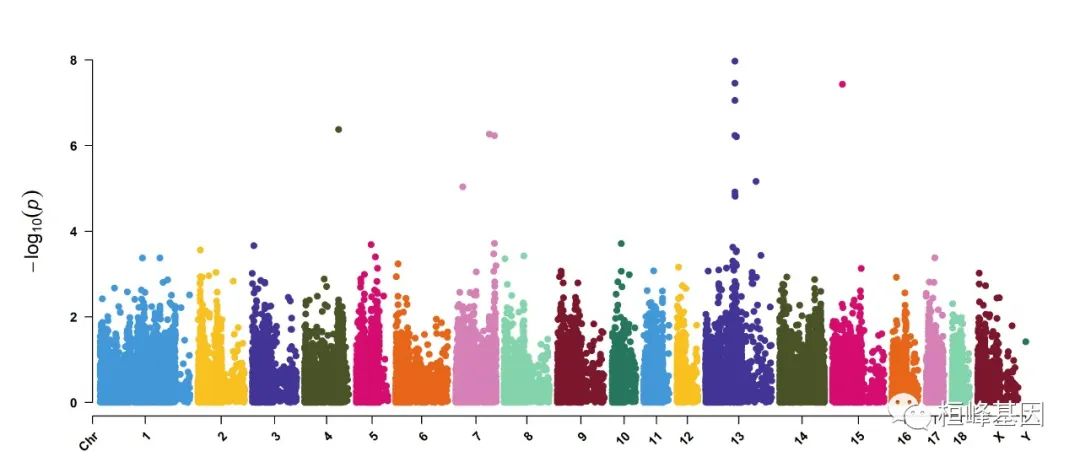
2. Amplify signals on pch, cex and col
CMplot(pig60K, plot.type="m", col=c("grey30","grey60"), LOG10=TRUE, ylim=c(2,12), threshold=c(1e-6,1e-4),
threshold.lty=c(1,2), threshold.lwd=c(1,1), threshold.col=c("black","grey"), amplify=TRUE,
chr.den.col=NULL, signal.col=c("red","green"), signal.cex=c(1.5,1.5),signal.pch=c(19,19),
file="jpg",memo="",dpi=300,file.output=TRUE,verbose=TRUE,width=14,height=6)
3. Attach chromosome density on the bottom of Manhattan plot
CMplot(pig60K, plot.type = "m", LOG10 = TRUE, ylim = NULL, threshold = c(1e-06, 1e-04),
threshold.lty = c(1, 2), threshold.lwd = c(1, 1), threshold.col = c("black",
"grey"), amplify = TRUE, bin.size = 1e+06, chr.den.col = c("darkgreen", "yellow",
"red"), signal.col = c("red", "green"), signal.cex = c(1.5, 1.5), signal.pch = c(19,
19), file = "jpg", memo = "", dpi = 300, file.output = TRUE, verbose = TRUE,
width = 14, height = 6)
## Rectangular-Manhattan Plotting trait1.
## Rectangular-Manhattan Plotting trait2.
## Rectangular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot



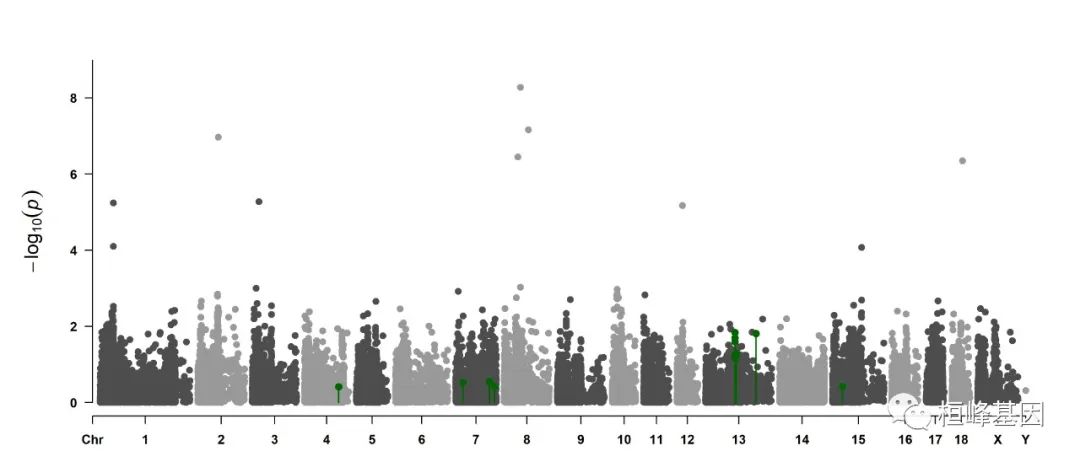
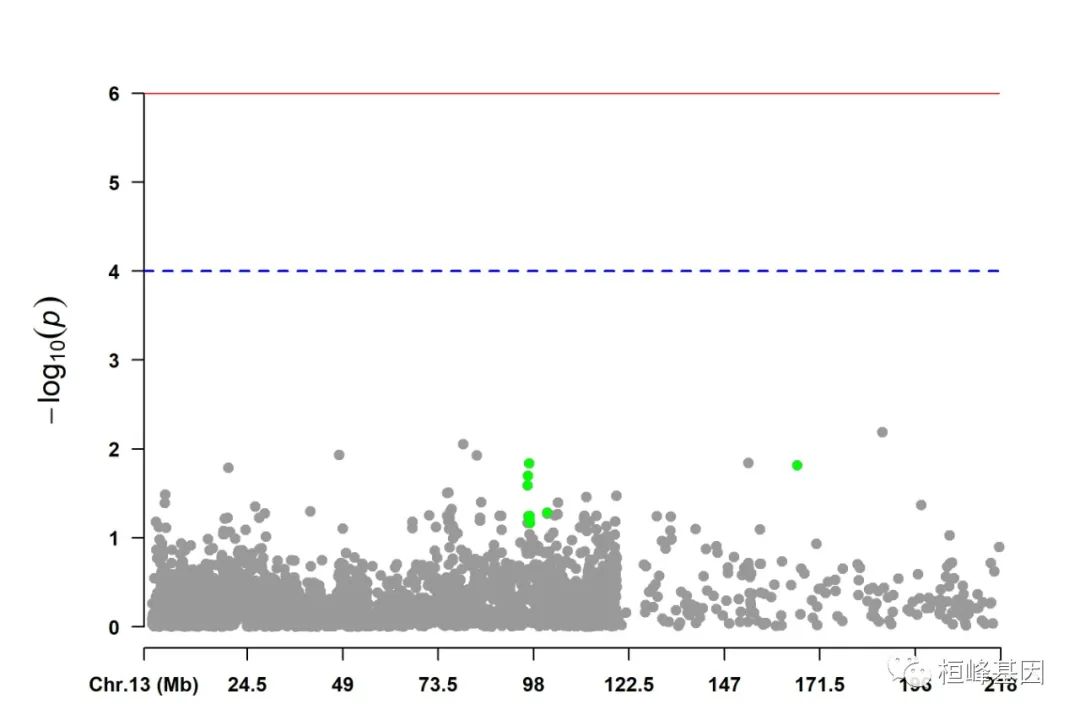
4. Highlight a group of SNPs on pch, cex, type, and col
signal <- pig60K$Position[which.min(pig60K$trait2)]
SNPs <- pig60K$SNP[pig60K$Chromosome == 13 & pig60K$Position < (signal + 1e+06) &
pig60K$Position > (signal - 1e+06)]
CMplot(pig60K, plot.type = "m", LOG10 = TRUE, col = c("grey30", "grey60"), highlight = SNPs,
highlight.col = "green", highlight.cex = 1, highlight.pch = 19, file = "jpg",
memo = "", chr.border = TRUE, dpi = 300, file.output = TRUE, verbose = TRUE,
width = 14, height = 6)
## Rectangular-Manhattan Plotting trait1.
## Rectangular-Manhattan Plotting trait2.
## Rectangular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplo
SNPs <- pig60K[pig60K$trait2 < 1e-04, 1]
CMplot(pig60K, type = "h", plot.type = "m", LOG10 = TRUE, highlight = SNPs, highlight.type = "p",
highlight.col = NULL, highlight.cex = 1.2, highlight.pch = 19, file = "jpg",
memo = "", dpi = 300, file.output = TRUE, verbose = TRUE, width = 14, height = 6,
band = 0.6)
## Rectangular-Manhattan Plotting trait1.
## Rectangular-Manhattan Plotting trait2.
## Rectangular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot
SNPs <- pig60K[pig60K$trait2 < 1e-04, 1]
CMplot(pig60K, type = "p", plot.type = "m", LOG10 = TRUE, highlight = SNPs, highlight.type = "h",
col = c("grey30", "grey60"), highlight.col = "darkgreen", highlight.cex = 1.2,
highlight.pch = 19, file = "jpg", dpi = 300, file.output = TRUE, verbose = TRUE,
width = 14, height = 6)
## Rectangular-Manhattan Plotting trait1.
## Rectangular-Manhattan Plotting trait2.
## Rectangular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot
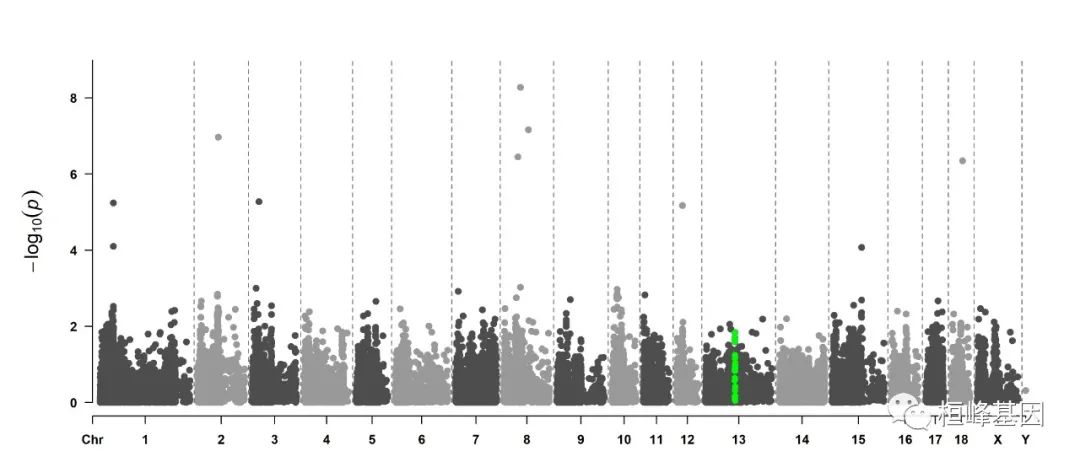
5. Visualize only one chromosome
CMplot(pig60K[pig60K$Chromosome == 13, ], plot.type = "m", LOG10 = TRUE, col = c("grey60"),
highlight = SNPs, highlight.col = "green", highlight.cex = 1, highlight.pch = 19,
file = "jpg", memo = "", threshold = c(1e-06, 1e-04), threshold.lty = c(1, 2),
threshold.lwd = c(1, 2), width = 9, height = 6, threshold.col = c("red", "blue"),
amplify = FALSE, dpi = 300, file.output = TRUE, verbose = TRUE)
## Rectangular-Manhattan Plotting trait1.
## Rectangular-Manhattan Plotting trait2.
## Rectangular-Manhattan Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot
6. add genes or SNP names around the highlighted SNPs
SNPs <- pig60K[pig60K[, 5] < (0.05/nrow(pig60K)), 1]
genes <- paste("GENE", 1:length(SNPs), sep = "_")
set.seed(666666)
CMplot(pig60K[, c(1:3, 5)], plot.type = "m", LOG10 = TRUE, col = c("grey30", "grey60"),
highlight = SNPs, highlight.col = c("red", "blue", "green"), highlight.cex = 1,
highlight.pch = c(15:17), highlight.text = genes, highlight.text.col = c("red",
"blue", "green"), threshold = 0.05/nrow(pig60K), threshold.lty = 2, amplify = FALSE,
file = "jpg", memo = "", dpi = 300, file.output = TRUE, verbose = TRUE, width = 14,
height = 6)
## Rectangular-Manhattan Plotting trait2.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot

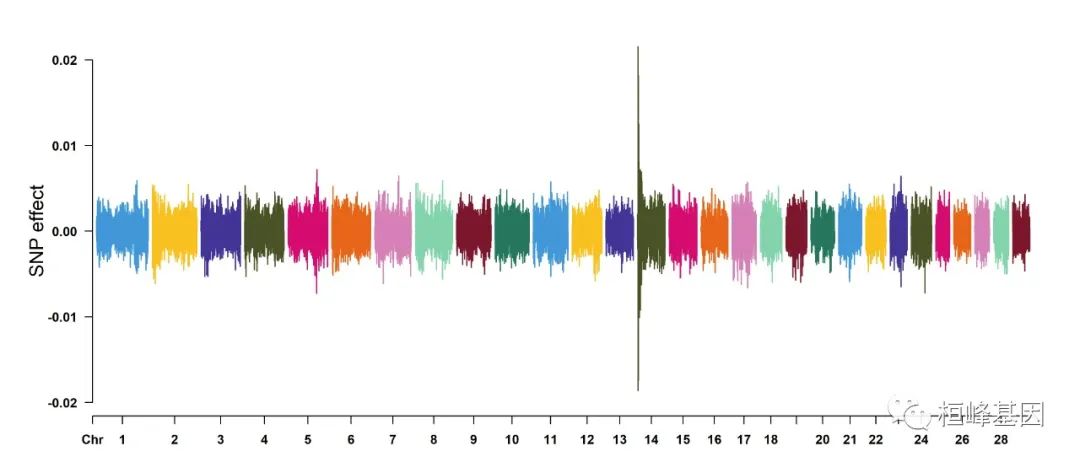
7. Genomic Selection/Prediction(GS/GP) or other none p-values
CMplot(cattle50K, plot.type = "m", band = 0.5, LOG10 = FALSE, ylab = "SNP effect",
threshold = 0.015, threshold.lty = 2, threshold.lwd = 1, threshold.col = "red",
amplify = TRUE, width = 14, height = 6, signal.col = NULL, chr.den.col = NULL,
file = "jpg", memo = "", dpi = 300, file.output = TRUE, verbose = TRUE, cex = 0.8)
## Rectangular-Manhattan Plotting Somatic cell score.
## Rectangular-Manhattan Plotting Milk yield.
## Rectangular-Manhattan Plotting Fat percentage.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot
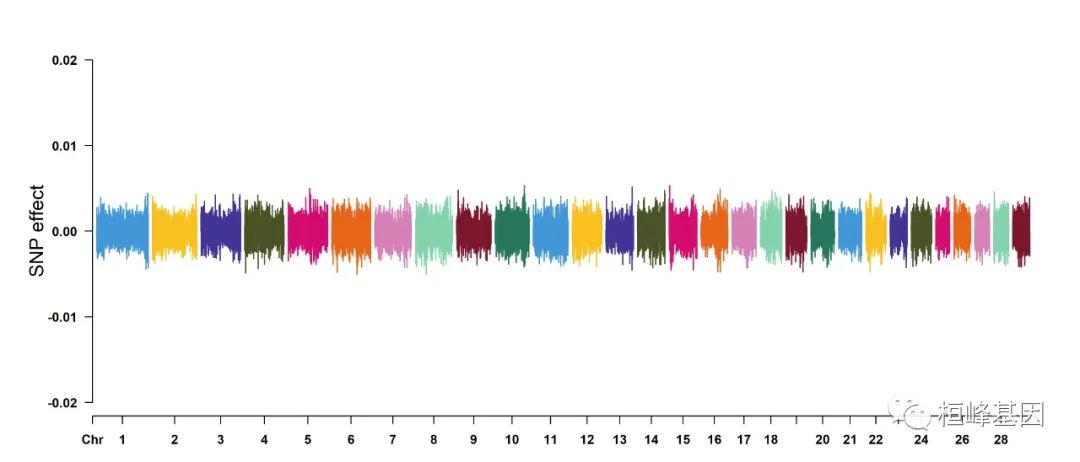
cattle50K[, 4:ncol(cattle50K)] <- apply(cattle50K[, 4:ncol(cattle50K)], 2, function(x) x *
sample(c(1, -1), length(x), rep = TRUE))
CMplot(cattle50K, type = "h", plot.type = "m", band = 0.5, LOG10 = FALSE, ylab = "SNP effect",
ylim = c(-0.02, 0.02), threshold.lty = 2, threshold.lwd = 1, threshold.col = "red",
amplify = FALSE, cex = 0.6, chr.den.col = NULL, file = "jpg", memo = "", dpi = 300,
file.output = TRUE, verbose = TRUE)
## (warning: all phenotypes will use the same ylim.)
## Rectangular-Manhattan Plotting Somatic cell score.
## Rectangular-Manhattan Plotting Milk yield.
## Rectangular-Manhattan Plotting Fat percentage.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot


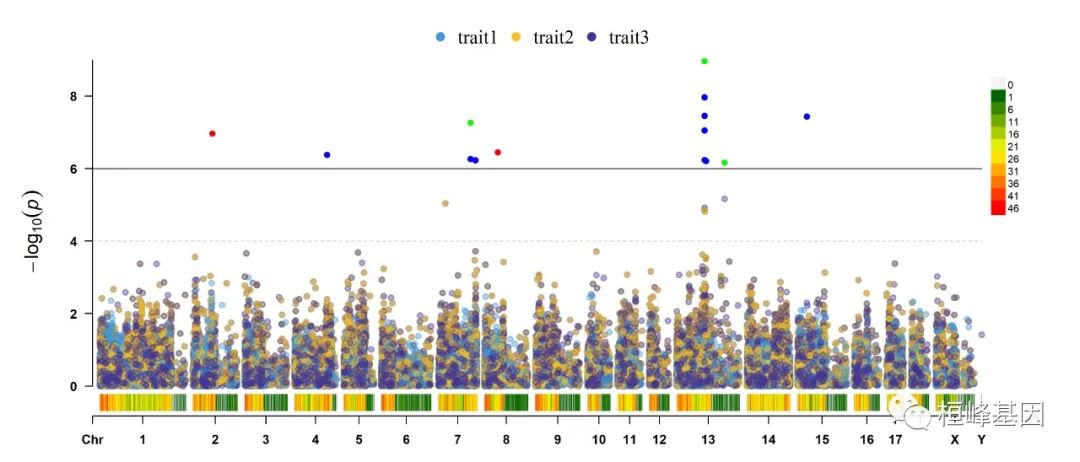
4. Multi_tracks Rectangular-Manhattan plot
SNPs <- list(pig60K$SNP[pig60K$trait1 < 1e-06], pig60K$SNP[pig60K$trait2 < 1e-06],
pig60K$SNP[pig60K$trait3 < 1e-06])
CMplot(pig60K, plot.type = "m", multracks = TRUE, threshold = c(1e-06, 1e-04), threshold.lty = c(1,
2), threshold.lwd = c(1, 1), threshold.col = c("black", "grey"), amplify = TRUE,
bin.size = 1e+06, chr.den.col = c("darkgreen", "yellow", "red"), signal.col = c("red",
"green", "blue"), signal.cex = 1, file = "jpg", memo = "", dpi = 300, file.output = TRUE,
verbose = TRUE, highlight = SNPs, highlight.text = SNPs, highlight.text.cex = 1.4)
## Multracks-Manhattan Plotting trait1.
## Multracks-Manhattan Plotting trait2.
## Multracks-Manhattan Plotting trait3.
## Multraits-Rectangular Plotting...(finished 13%)
## Plots are stored in: F:/demo script/基因组分析系列/CMplot
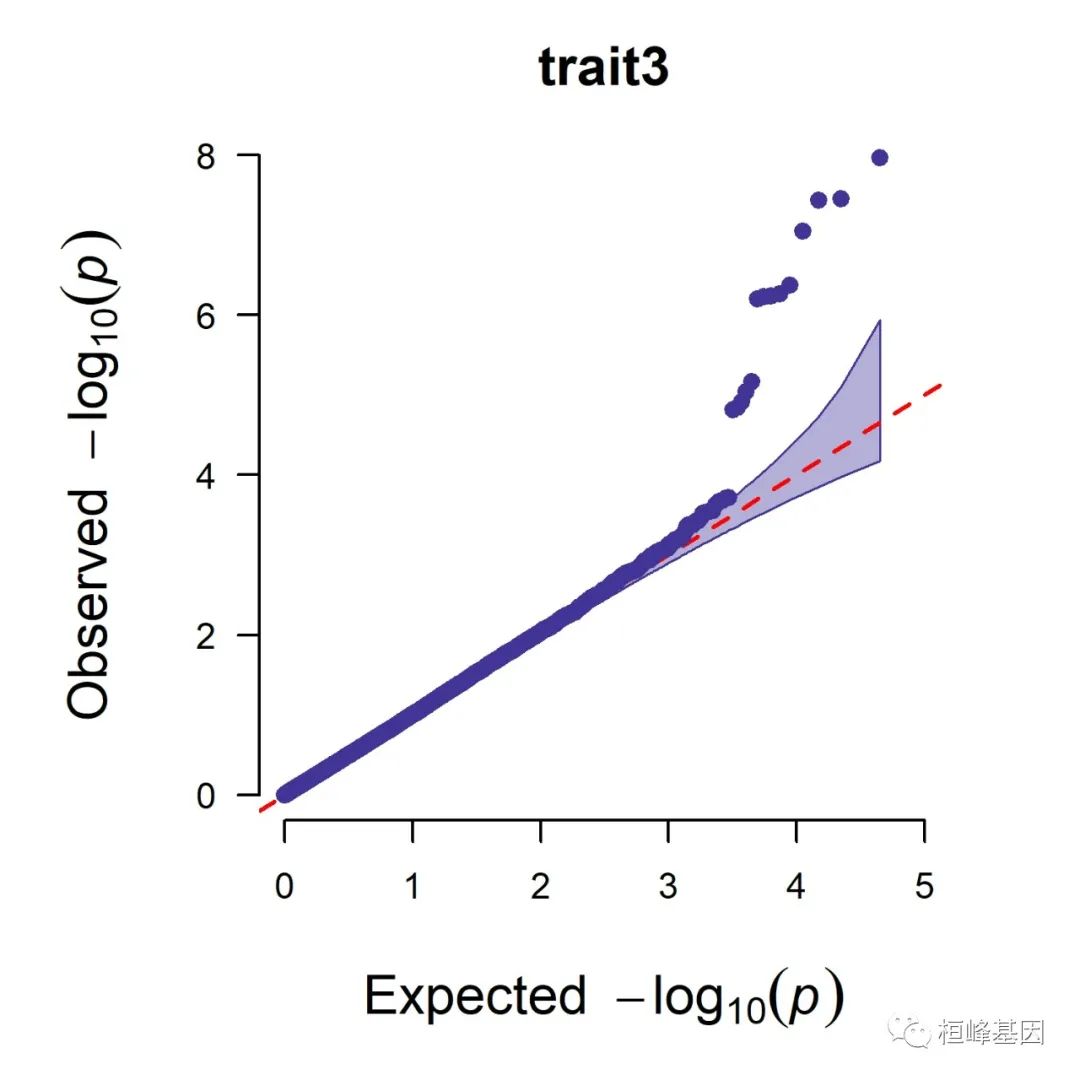
5. Q-Q plot
1. Single_track Q-Q plot
CMplot(pig60K, plot.type = "q", box = FALSE, file = "jpg", memo = "", dpi = 300,
conf.int = TRUE, conf.int.col = NULL, threshold.col = "red", threshold.lty = 2,
file.output = TRUE, verbose = TRUE, width = 5, height = 5)
## QQ Plotting trait1.
## QQ Plotting trait2.
## QQ Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot
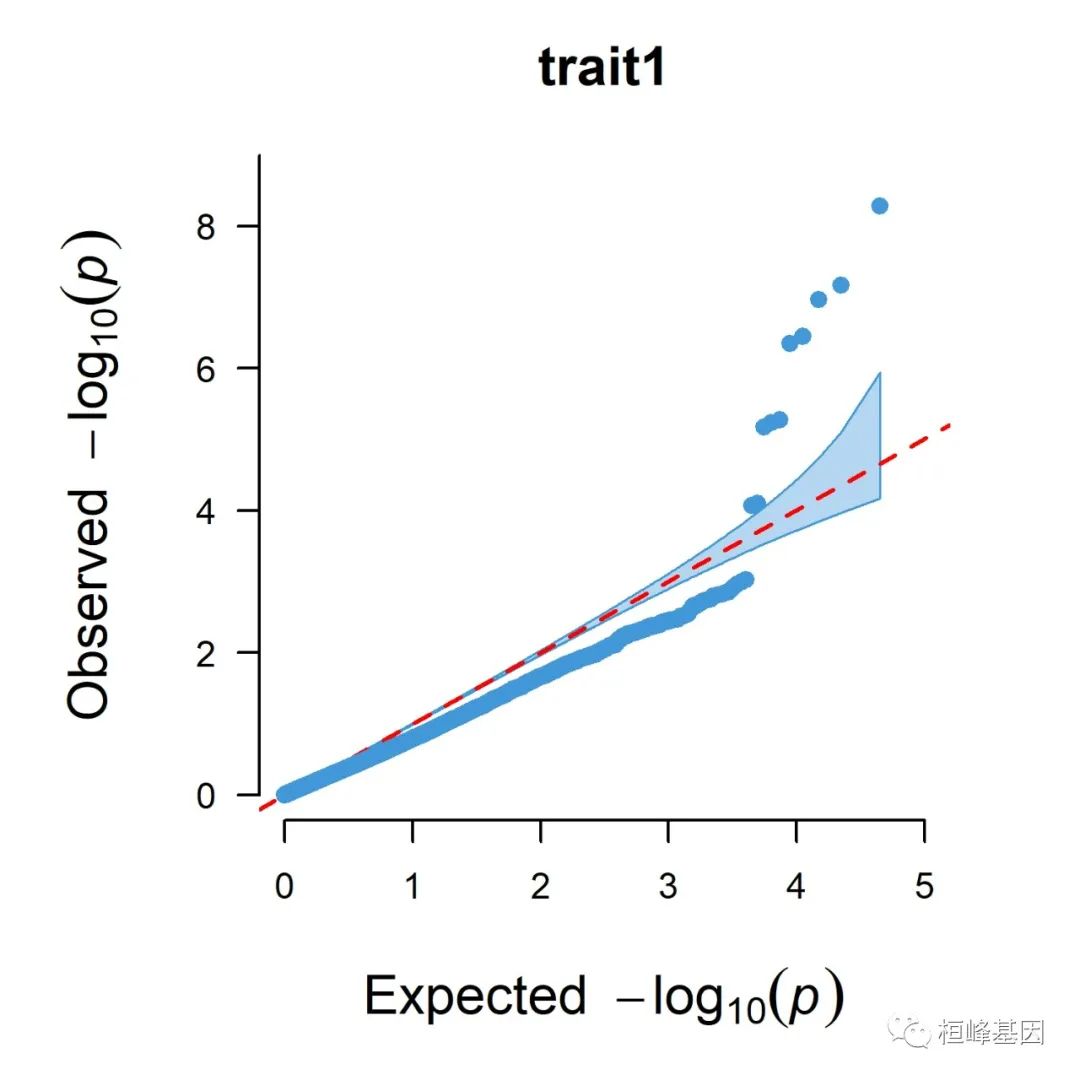
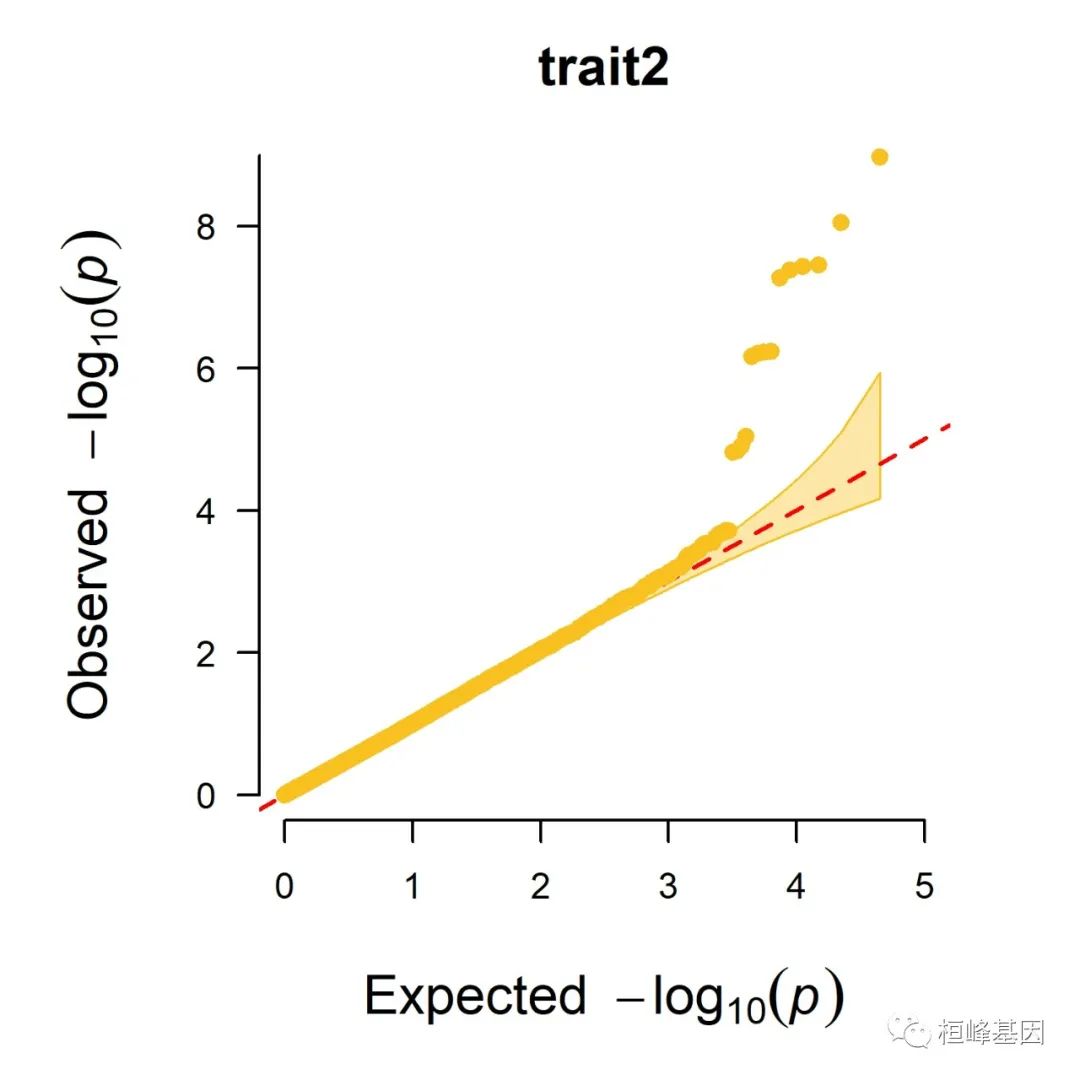
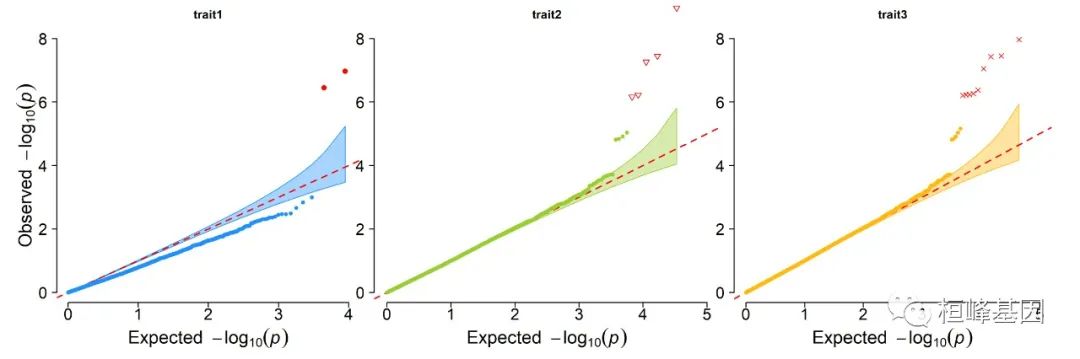
2. Multi_tracks Q-Q plot
pig60K$trait1[sample(1:nrow(pig60K), round(nrow(pig60K) * 0.8))] <- NA
pig60K$trait2[sample(1:nrow(pig60K), round(nrow(pig60K) * 0.25))] <- NA
CMplot(pig60K, plot.type = "q", col = c("dodgerblue1", "olivedrab3", "darkgoldenrod1"),
threshold = 1e-06, ylab.pos = 2, signal.pch = c(19, 6, 4), signal.cex = 1.2,
signal.col = "red", conf.int = TRUE, box = FALSE, multracks = TRUE, cex.axis = 2,
file = "jpg", memo = "", dpi = 300, file.output = TRUE, verbose = TRUE, ylim = c(0,
8), width = 5, height = 5)
## (warning: all phenotypes will use the same ylim.)
## Multracks-QQ Plotting trait1.
## Multracks-QQ Plotting trait2.
## Multracks-QQ Plotting trait3.
## Multraits-QQ Plotting trait1.
## Multraits-QQ Plotting trait2.
## Multraits-QQ Plotting trait3.
## Plots are stored in: F:/demo script/基因组分析系列/CMplot

References:
Yin, L. et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated tool for Genome-Wide Association Study, Genomics, Proteomics & Bioinformatics (2021), doi: 10.1016/j.gpb.2020.10.007.

以上是关于GWAS基本分析内容的主要内容,如果未能解决你的问题,请参考以下文章