全基因组关联分析(Genome-Wide Association Study,GWAS)流程
Posted 橙子牛奶糖
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了全基因组关联分析(Genome-Wide Association Study,GWAS)流程相关的知识,希望对你有一定的参考价值。
全基因组关联分析流程:
一、准备plink文件
1、准备PED文件
PED文件至少有六列,内容如下:
Family ID
Individual ID
Paternal ID
Maternal ID
Sex (1=male; 2=female; other=unknown)
Phenotype(-9 missing 0 missing 1 unaffected 2 affected)
genotype( 1,2,3,4 or A,C,G,T missing 0)
PED文件是空格(空格或制表符)分隔的文件。
PED文件长这个样:

2、准备MAP文件
MAP文件有四列,四列内容如下:
chromosome (1-22, X, Y or 0 if unplaced)
rs# or snp identifier
Genetic distance (morgans)
Base-pair position (bp units)
MAP文件长这个样:
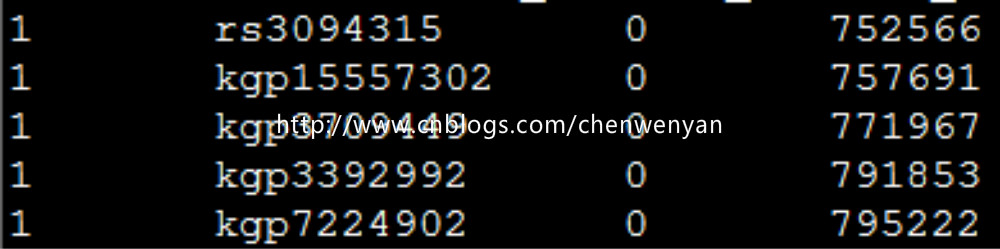
3、生成bed、fam、bim、文件
输入命令
plink --file mydata --out mydata --make-bed
注:plink指的是plink软件,如果软件安装在某个指定的路径的话,前面还要加上路径,比如安装在路径为/your/pathway/的文件夹下,则命令应该为“/your/pathway/plink --file mydata --out mydata --make-bed”
mydata指的是1和2生成的PED和MAP文件名,不需要写.ped和.map后缀
二、准备表型文件(Alternate phenotype files)
一般表型文件为txt格式,表型文件有三列,分别为:
Family ID
Individual ID
Phenotype
假如有多种表型,第一列和第二列还是Family ID、Individual ID,第三列及以后的每列都是表型,例如以下:
Family ID
Individual ID
Phenotype A
Phenotype B
Phenotype C
Phenotype D
Phenotype E
……
表型文件长这样:
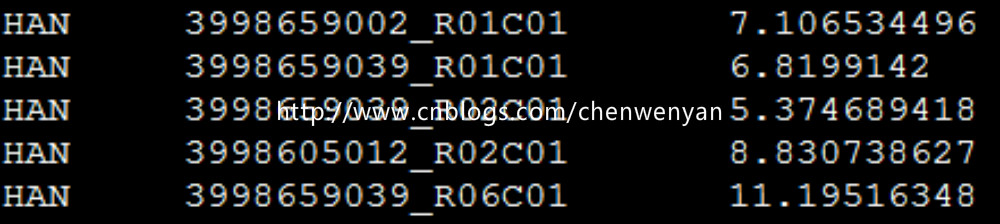
缺失值的处理:缺失值的表型用-9表示;
case和control的处理:通常情况下,1表示control,2表示case,0表示缺失,但如果你加上--1的参数,则0表示control,1表示case。
三、准备协变量文件(Covariate files)
协变量文件同表型文件类似,第一列和第二列是Family ID、Individual ID,第三列及以后的每列都是协变量
Family ID
Individual ID
Covariate A
Covariate B
Covariate C
Covariate D
Covariate E
……
协变量文件长这个样(这里有三个协变量,分别为Sex,Age,temperature):
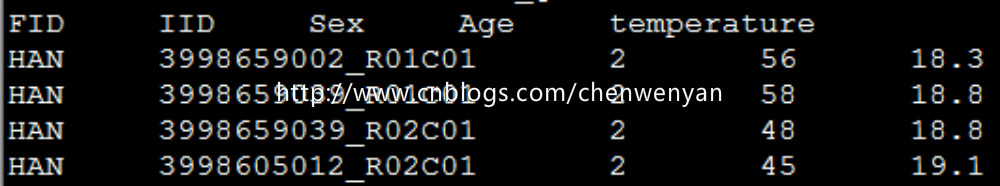
四、plink进行表型和基因型以及协变量的关联分析
命令如下:
plink --bfile mydata --linear --pheno pheno.txt --mpheno 1 --covar covar.txt --covar-number 1,2,3 --out mydata –noweb
生成的文件为mydata.assoc.linear
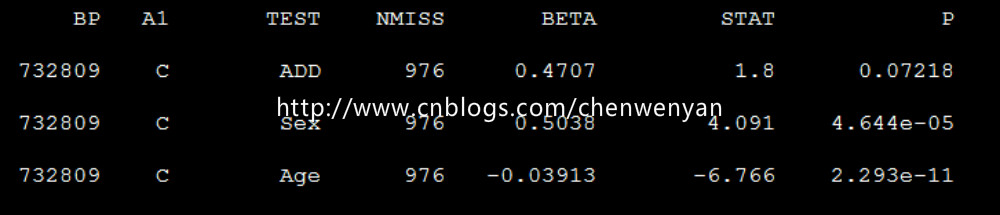
注:“mydata”mydata文件不需要后缀,“--mpheno 1”指的是表型文件的第三列(即第一个表型)
“--covar-number 1,2,3”指的是协变量文件的第三列、第四列、第五列(即第一个、第二个、第三个协变量)
“--linear”指的是用的连续型线性回归,如果表型为二项式(即0、1)类型,则用“--logistic”
五、画曼哈顿图
安装R语言的CpGassoc包,其中的manhattan(),即可画曼哈顿图,或者参照本文R语言画全基因组关联分析中的曼哈顿图(manhattan plot)
以上是关于全基因组关联分析(Genome-Wide Association Study,GWAS)流程的主要内容,如果未能解决你的问题,请参考以下文章