GWAS基本概念2
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GWAS基本概念2相关的知识,希望对你有一定的参考价值。
参考技术A 本篇主要说了LD连锁不平衡,LD的度量方法,直接关联和间接关联。原文还讲了很多其他知识,我这边并未全盘翻译。请移步原文。理解LD对于GWAS结果分析是非常重要的。在得到GWAS结果之前,我们用的是基因型和表型的文件,通过一定的软件,选择一定的统计学方法,得到关联分析的结果。这一切操作都不涉及LD这个概念。但是,后续筛选到的大量的SNP,如何找到致病基因,就需要应用LD这个原理了。文章来自:Chapter 11: Genome-Wide Association Studies
3.2 Linkage Disequilibrium
~
Linkage disequilibrium (LD) is a property of SNPs on a contiguous stretch of
genomic sequence that describes the degree to which an allele of one SNP is inherited or correlated with an allele of another SNP within a population
~
连锁不平衡(LD)是基因组序列连续片段上SNP的属性,其描述了一个SNP的等位基因在群体内遗传或与另一个SNP的等位基因相关的程度。连锁不平衡这一术语是由群体遗传学家创造的,他们试图用数学方法描述种群内随时间变化的遗传变异。它与染色体连锁的概念有关,即一个染色体上的两个标记在一个家族的世代中保持物理连接。在图2中,显示了两条创始人染色体(一条蓝色,一条橙色)。家系内的世代重组事件会导致染色体片段的断裂。这种效应在世代中被放大,在一个固定大小的种群中,在随机交配时,重复的随机重组事件会打断相邻染色体的片段(包含连接等位基因),直到最终群体中的所有等位基因都处于连接平衡或相互独立。因此,群体尺度上标记间的连锁称为连锁不平衡。
(左图时家系内的连锁概念。两个创始者的染色体的片段进行了重组事件,然后,又经过多代的染色体重组,最终后代中的染色体含有多个被打碎重组的片段。然而,有一部分区段无论从祖先还是目前,都是紧密连锁的。图中的红色部分。
再看右边图,在一个群体中,从第一代开始,创始者染色体上连锁的区段被重组事件打破,缩小。随着时间的推移,群体中染色体上的一对标记从连锁不平衡走向到连锁平衡,这是因为重组事件最终发生在染色体上的每一个可能的点之间)
LD的衰减率取决于多种因素,包括种群大小、种群中创始染色体的数量以及种群已经存在的世代数。因此,不同的人类亚群具有不同的LD程度和模式。非洲裔是LD最古老的祖先,由于该群体中重组事件的积累,LD的区域更小。欧洲人后裔和亚洲人后裔是由始创事件(从非洲人口中提取的染色体样本)创造的,它改变了始创染色体的数量、群体规模和群体的时代年龄。这些人口的平均LD区域比非裔群体大。(从这段我们也可以明白,非洲人的历史最早,经过的历史重组事件也最多,LD区域小。LD衰减程度最大,在这三个人群中)。
虽然已经提出了许多LD的测量方法,但它们最终都与两个等位基因(即双标记单倍型)的共出现频率与两个标记独立时的期望频率之间的差异有关。连锁不平衡的两种常用测量方法是D '和r2,如公式1和2所示。在这些方程,π ab是ab单倍型的频率,π a:是a等位基因的频率,π b:是b等位基因的频率。D’是一种与标记间重组事件相关的群体遗传学度量,其范围为0和1。D'值为0表示完全连锁平衡,这意味着在Hardy-Weinberg平衡原则下,两个标记之间频繁的重组和在统计学上具有独立性。D'为1表示完全LD,表示群体内两个标记之间没有重组。为了遗传分析的目的,LD通常是以r2(一种相关性的统计度量)来报告的。高r2值表明两个SNP传递相似的信息,因为第一个SNP的一个等位基因经常与第二个SNP的一个等位基因被观察到,所以只需要对两个SNP中的一个进行基因分型就可以捕获等位基因变异。这两个统计数据之间存在依赖关系;r2对两个标记的等位基因频率敏感,并且只能在高D '区域高。
与LD测量相关的一个经常被遗忘的问题是,目前的技术不允许直接测量样本中的单倍型频率,因为每个SNP都是独立的基因分型,而且每个等位基因的相位起源或染色体的起源是未知的。许多成熟的和有文献记载的方法用于推断单倍型相位和估计随后的双标记单倍型频率,一般会产生合理的结果。(这个意思好像和后面的基因型填补有关,基因型填补可以补充那些缺失的SNP标记。填充的推断原理应该就是根据连锁不平衡来的,现在有好几款软件可以用于人类和非人类)。
被特异性选择用来捕捉基因组附近位点变异的snp称为tag snp,因为这些snp的等位基因标记周围的LD延伸。如前所述,LD的模式是特定于群体的,因此,为一个群体选择的tag snp对于不同的群体可能不起作用。LD被用来优化遗传研究,防止基因型snp提供多余的信息。根据HapMap项目的数据分析,欧洲后裔群体中80%的常见单核苷酸多态性可以通过分布在基因组中的50万到100万个单核苷酸多态性的子集获得。(感觉没太看懂,需要搜点别的资料)
3.3 Indirect Association
由于LD的存在,在GWAS中产生了两种可能的积极结果。在第一个结果中,SNP影响一个生物系统,最终导致表型改变。这种情况下是直接进行基因分型的,并且统计上与性状相关。这被称为直接关联,而被分型的SNP有时也被称为功能性SNP(functional SNP)。第二种可能性是, influential SNP并没有被分型,但是,与 influential SNP相处在一个高LD区域中的tag SNP被分型,并在统计学上与表型相关(图3),这被称为间接关联。由于这两种可能性,GWAS结果中一个显著的SNP关联,不应该被假定为因果变异,并且可能需要额外的研究来确定influential SNP的精确位置。
从概念上讲,GWAS在常见疾病/常见变异假说下的最终结果是,一个由50万到100万个标记组成的小组将识别与常见表型相关的常见snp。要进行这样的研究,实际上需要一种基因分型技术,这种技术能够以一种有效的方式准确地捕获研究中每个个体的50万到100万个snp的等位基因。(翻译不准,大致意思就是需要一个高通量的基因分型技术,来获得大量的SNP标记)。
(灰色的点是基因分型得到的SNP,红色的是致病SNP。实际上,我们做GWAS的结果,很多不会得到我们想要的结果,即直接关联的SNP。我们通常由这个关联到的SNP,根据LD原理,在一个LD block区域内寻找真正的致病SNP)。
括号内容是我根据自己的理解加入的。请尽量翻看原文。看文章不要看一篇,多看看几篇就会理解了。
GWAS文献基于GWAS与群体进化分析挖掘大豆相关基因
发表期刊杂志:nature biotechnology
影响因子:41.514
发表时间:2015年2月
发表单位:中科院遗传与发育生物学研究所
一、 研究取材
62株野生大豆、130株地方种和110个驯化品种构建的一个自然群体
二、 方法流程
Illumina HiSeq 2000 测序平台,测序文库300bp,样本平均测序深度达到11X
三、 生物信息学分析
群体结构分析、选择清除分析、重要性状的全基因组关联分析
四、 研究结果
1)使用BWA软件将原始数据与参考基因组进行比对,使用samtools将sam格式转化为bam,使用picard软件去掉Duplicated reads。
2)SNP calling使用GATK和samtools,取两者结果的交集。对于GATK参数设置:-stand_call_conf 30。MAF设置为0.01。
3) Indel calling类似于SNP calling,使用GATK的UnifiedGenotyper程序,起参数设置为-glm INDEL,只考虑6bp范围内的缺失和插入。
4)SNP注释使用的软件为ANNOVAR。SNP被注释到内含子(overlap- ping with an intron)、外显子、基因间区,可变剪切位点(within 2 bp of a splicing junction)、5′UTRs 、3′UTRs,, upstream and downstream regions (within a 1 kb region upstream or downstream from the transcription start site).注释在外显子区域的SNP又分为同义和非同义突变。注释到外显子的Indel又分为移码突变和非移码突变。
5)群体结构分析中,PCA使用的是EIGENSOFT 4.2 的smartpca 程序,neighbor-joining tree 使用PHYLIP 3.68软件。结构分层使用FRAPPE,其中k值选取2到7.连锁不平衡分析使用plink软件。关联分析使用的GAPIT 分析软件。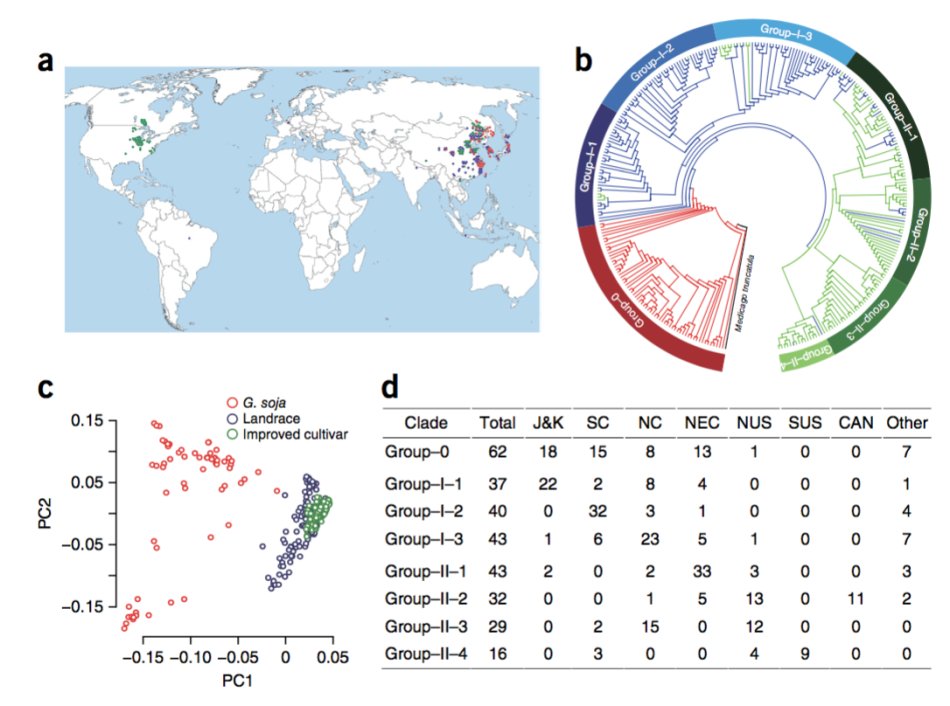
以上是关于GWAS基本概念2的主要内容,如果未能解决你的问题,请参考以下文章