序列比对及算法模式的选择
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了序列比对及算法模式的选择相关的知识,希望对你有一定的参考价值。
参考技术A 选择序列比对的方法:Muscle或者ClustalW。 ClustalW 的基本原理是首先做序列的两两比对,根据该两两比对计算两两距离矩阵,是一种经典的比对方法,使用范围也比较广泛。 Muscle 的功能仅限于多序列比对,它的最大优势是速度,比ClustalW的速度快几个数量级,而且序列数越多速度的差别越大。方法可以通过点击图中上方Alignment或者下方的图标「W」和「Muscle」来选择。如果你的序列是 DNA编码序列 ,就一定要选择 Align Codons ,因为序列通过密码子比对比DNA序列的比对会更加真实,避免间隙对比对结果产生的影响。
(但是该选项会因为序列中含有终止密码而报错。)
比对之后要去检查一下比对的情况,有的差异很大的或许是因为序列方向反了,这个时候要把它反转回来,右击这条序列,点击Reverse Complement,反转后一定要再次点击比对,检查是否大部分都对齐了。
分析后返回主页面,接下来我们要选择一个最优的模型,提高建树的精确度。点击 MODELS 中的 Find Best DNA/Protein Models(ML) 软件就会根据你的数据帮你计算寻找最适合的模型。
分析计算结果。具有 最低BIC分数 (BayesianInformation Criterion)的模型被认为 是最好地描述替代模式 。对于每个模型,还给出了AICc值(Akaike Information Criterion, corrected,值越低拟合程度越好),以及用来计算上述两个分值的最大似然值(lnL)和参数数量(包括分支长度)。在这里就可以看到,BIC分数最低的模型是K2+G+I,K2+G+I在这里就是最好的模型。
但因为实际在后面的模型选择中, 软件有时没有提供组合的模型来选择 ,所以我们继续看下面的BIC分数,可以找到 单个模型 中得分 最小的 ,就是我们在这里要选择的最优模型。
序列比对和构建进化树(clustalw和phylip)
安装clustalw很简单,不提了。
找了几个蛋白序列进行比对,命名为dm.fasta
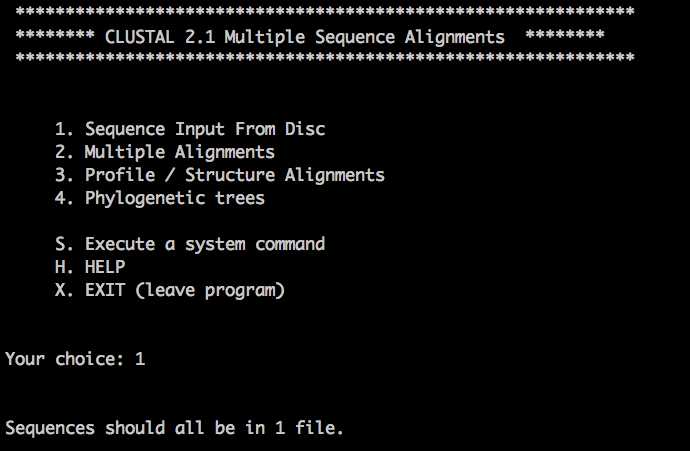
1、输入 ./clustalw2 进入交互模式
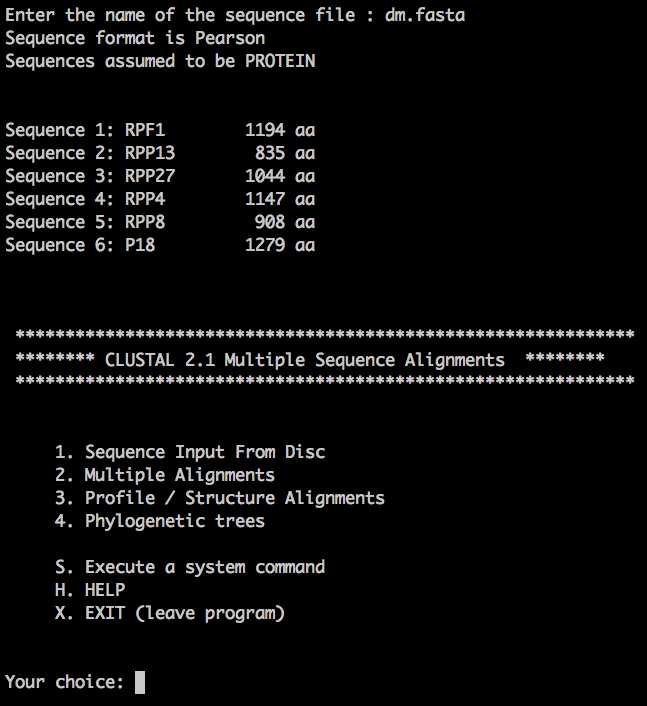
2、选择1 并输入文件名字
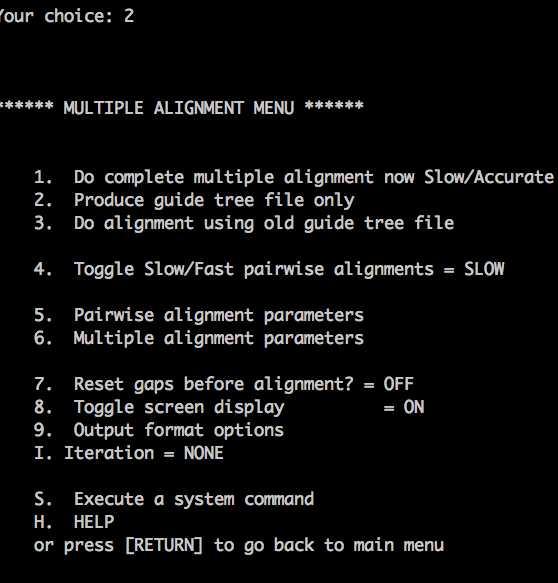
3、输入2, 进行多序列比对
4、如果要修改输入格式,则点9

5、若要输出格式为phylip,则点4,并关闭1
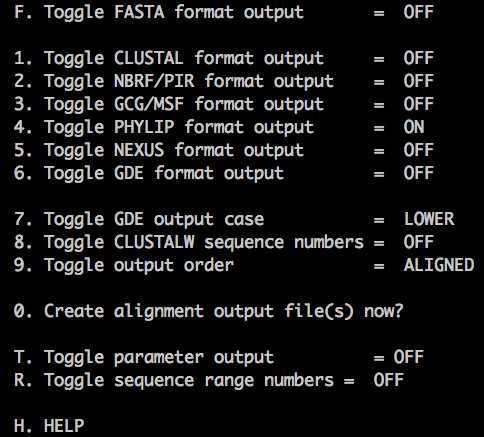
6、按下回车,后退
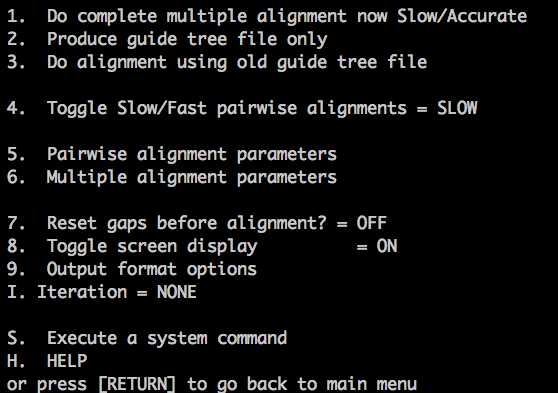
7、选择1进行比对, 因为phylip输入文件为名infile, 所以这里直接改名字infile,并退出软件即可
安装phylip
减压后,进入src 并输入 make -f Makefile.unx install
然后进入exe, 并将刚才比对结果infile 移到exe中。
进行构建树:
1、最大似然
直接输入./proml , 输入y进行确定参数,得到两个文件,outtree 和outfile,若想图形化,则将outtree 改为outtree.tre 可在mega上查看
2、临接法
先输入./protdist. 计算各个序列中两两序列的距离,得到距离矩阵。将该结果文件改名为infile,并进行临接法构进化树,方法为;输入./neighbor.用同样的方法可以在mega上查看图像
以上是关于序列比对及算法模式的选择的主要内容,如果未能解决你的问题,请参考以下文章