R语言整理TCAG编码RNA数据
Posted 易学统计
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言整理TCAG编码RNA数据相关的知识,希望对你有一定的参考价值。
已经被小伙伴们催更了,最近实在是太忙了,不过更新这件事,一直在路上。后台也收到许多留言,感谢大家的提问。这次介绍的是如何用R语言来处理从TCGA上下载编码RNA的数据。
在上次保存下载数据的文件中,新建一个文件夹基因数据,然后选择已下载的基因数据 gdc_download_20200728_060020.290637.tar ,鼠标右键选择解压到 基因数据 这个文件夹。
这个地方给大家截图的原因是后面文件夹够多,以免混乱。
打开基因数据这个文件夹 可以看到里面有182个文件夹,每一个文件夹里面还有1个压缩文件。这里面其实就是下载的182个样本,每个样本的信息单独保存在一个文件夹。这种数据不能直接拿来做分析,因此先要合并。
在基因数据这个文件中新建一个文件夹 00_mobile_data_to_one_file
利用R中的复制功能,将这182个文件夹中的压缩文件全部提取,并统一保存到00_mobile_data_to_one_file 这个文件夹。这个文件夹为啥要以00开头呢,是因为00开头就可以将该文件夹置顶,便于查找,就这意思。
setwd('~\下载数据\基因数据\')Dir <- dir()for (dirname in dir()[2:183]){file <- list.files(dirname,pattern = "*.counts")file.copy(paste0(dirname,"/",file),"00_mobile_data_to_one_file")}
运行完了之后,我们可以打开文件夹看一下,确实在里面,这时候可以全部选择,将文件解压到gene_data_unzip文件夹。
setwd('~\下载数据')#读入json格式的文件,他是一个182行,14列的数据框metadata <- jsonlite::fromJSON("metadata.cart.2020-07-28.json")#转换的就是两列file_name和associated_entities,我们把它选出来library(dplyr)metadata_id <- metadata %>%select(c(file_name,associated_entities))
注意第一点:这里可通过json的在线编辑器看下文件的内容
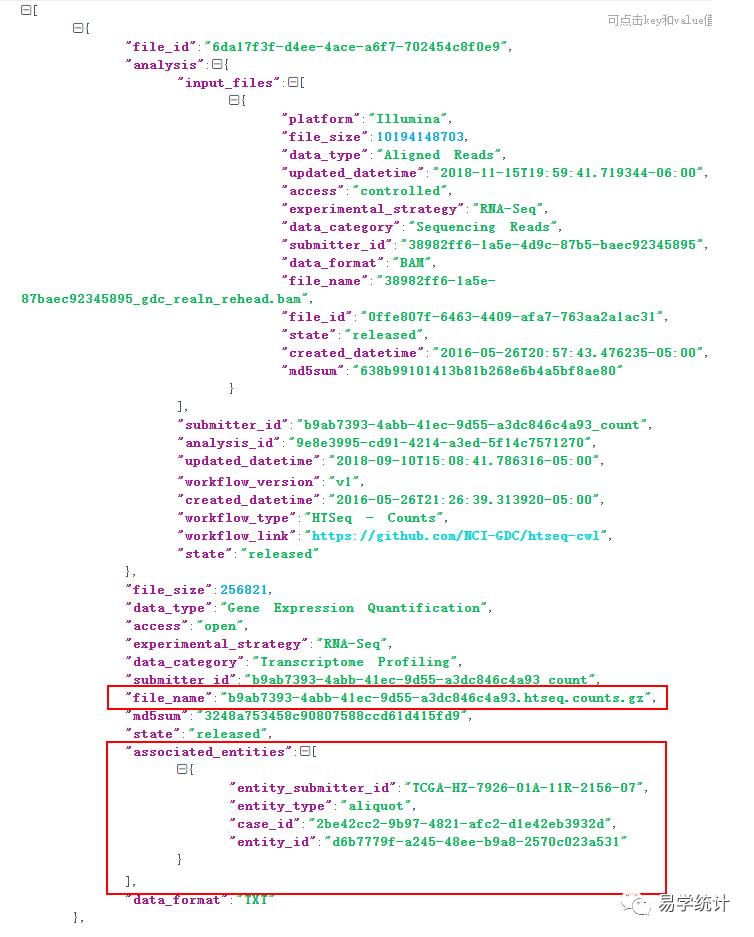
第一个红框对应的是基因文件的名称。
第二个红框是对应样本名称。
根据这个表就可以将基因文件和样本名称对应起来,形成一个大表。
根据上面的分析,只需要file_name和associated_entities中的entity_submitter_id,写R代码提取出来。
#做成一个数据框,然后我批量对应转换id <- data.frame()for (i in 1:182){id[i,1] <- substr(metadata_id$file_name[i],1,nchar(metadata_id$file_name[i])-3)id[i,2] <- metadata_id$associated_entities[i][[1]]$entity_submitter_id}
library(data.table)nameList <- list.files("gene_data_unzip/")location <- which(id==nameList[1],arr.ind = TRUE)TCGA_id <- as.character(id[location[1],2])expr_df<- fread(paste0("gene_data_unzip/",nameList[1]))names(expr_df) <- c("gene_id",TCGA_id)for (i in 2:length(nameList)){location <- which(id==nameList[i],arr.ind = TRUE)TCGA_id <- as.character(id[location[1],2])dfnew <- fread(paste0("gene_data_unzip/",nameList[i]))names(dfnew) <- c("gene_id",TCGA_id)expr_df <- inner_join(expr_df,dfnew,by="gene_id")}
总共60488行,查看最后几行发现有5行不是我们要的,去掉最后五行
expr_df <- expr_df[1:(length(expr_df$gene_id)-5),]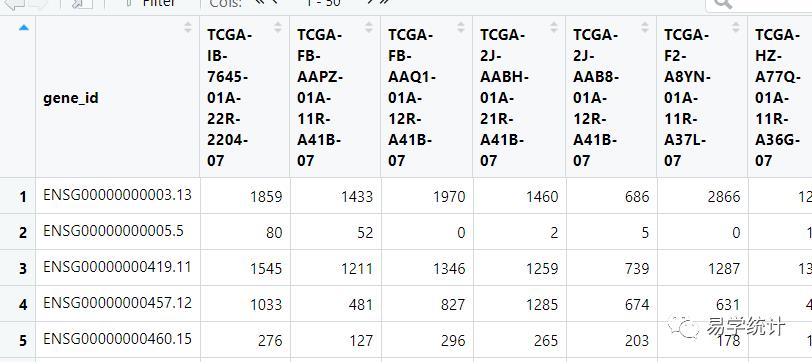
如上图的表的第一列是基因的ID,现在需要根据基因注释文件,将这个ID转化成与之对应的基因名称,这里找到两个基因注释的文件,分别做对应,具体这两个基因注释文件有什么区分,感兴趣的小伙伴可以深入研究下。
方法一:从这个网址上下载GTF文件,点击即可下载并解压。
ftp://ftp.ensembl.org/pub/release-99/gtf/homo_sapiens
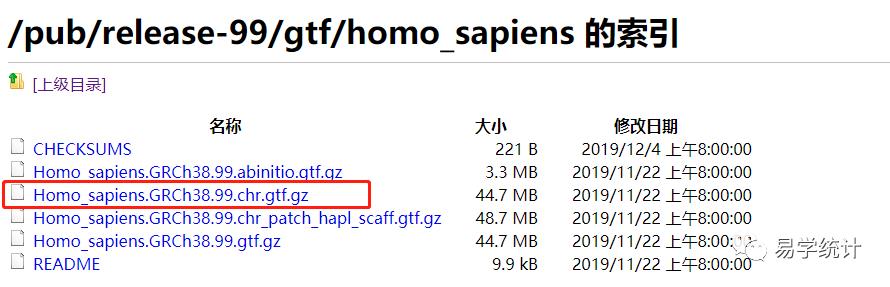
##需要安装这两个包读取gtf数据。library(SummarizedExperiment)library(rtracklayer)gtf1 <- rtracklayer::import('Homo_sapiens.GRCh38.99.chr.gtf')###需要几分钟gtf_df <- as.data.frame(gtf1)##保存数据save(gtf_df,file = "gtf_df.Rda")view(head(gtf_df))
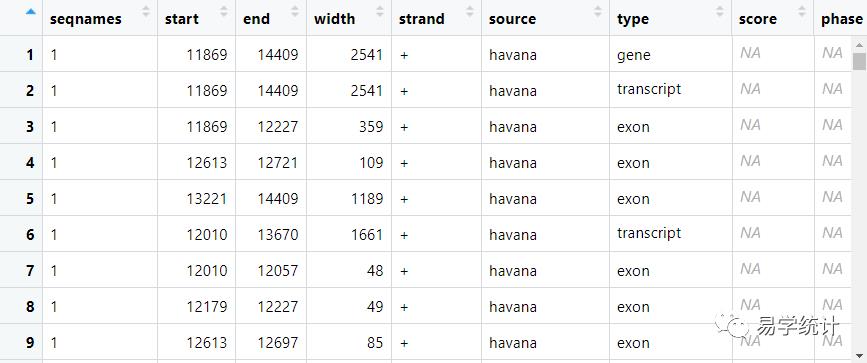
这个表里面的具体信息可以查看:https://www.jianshu.com/p/7cec5a02768a
关键一步来了,合并数据操作:
library(dplyr)library(tidyr)expr_df_nopoint <- expr_df %>%tidyr::separate(gene_id,into = c("gene_id","num"),sep="\.") %>%dplyr::select(-num)save(expr_df_nopoint,file = "expr_df_nopoint.Rda")
如何提取mRNA的表达矩阵?以gtf表(如上图所示)中的gene_biotype这个字段等于"protein_coding",且 字段 type等于"gene",筛选出来的基因就是编码基因。R代码如下:
library(dplyr)library(tidyr)mRNA_exprSet <- gtf_df %>%dplyr::filter(type=="gene",gene_biotype=="protein_coding") %>% #筛选gene,和编码指标dplyr::select(c(gene_name,gene_id,gene_biotype)) %>%dplyr::inner_join(expr_df_nopoint,by ="gene_id") %>%tidyr::unite(gene_id,gene_name,gene_id,gene_biotype,sep = " | ")
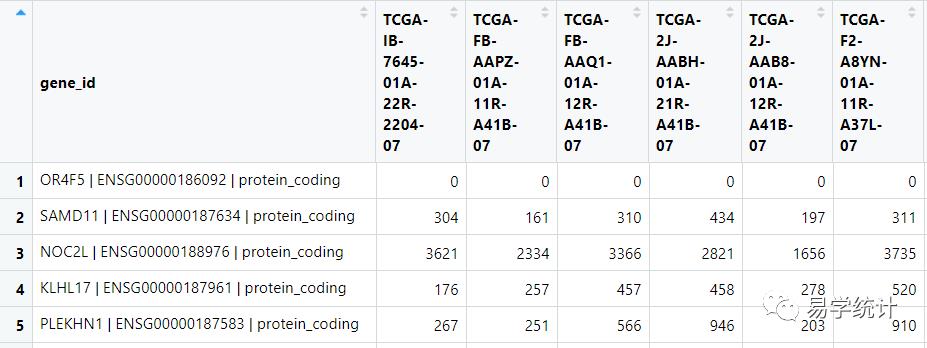
这个整合完的表有19668个基因,接下来这个表就可以进行差异分析了。
方法二:采用R封装的一个包进行基因ID转换
##首先安装这两个包,第一个是数据整合包,第二个是内置的基因注释包library(clusterProfiler)library(org.Hs.eg.db)###ids <- bitr(expr_df_nopoint$gene_id, fromType="ENSEMBL",toType=c("SYMBOL"), OrgDb="org.Hs.eg.db", drop=F)###去掉空值ids <- ids[!is.na(ids$SYMBOL),]###colnames(ids) <- c('gene_id','gene_name')mRNA_Set <- expr_df_nopoint %>%dplyr::inner_join(ids,by ="gene_id") %>%tidyr::unite(gene_id,gene_name,gene_id,sep = " | ")
这个整合完的表有25880个基因,接下来这个表就可以进行差异分析。这两个方法都可以对应基因名称,但对应完的数量不太一样,感兴趣的朋友可以对比深度挖掘下。
已经形成基因表达的矩阵了,后面就是如何做差异表达,如有意愿,可继续关注。
作者介绍:医疗大数据统计分析师,擅长R语言。
欢迎各位在后台留言,恳请斧正!
更多阅读
长按二维码
易学统计
以上是关于R语言整理TCAG编码RNA数据的主要内容,如果未能解决你的问题,请参考以下文章
收藏 | 给你一份R语言入门教程,拜托你快点学习R语言吧!要不然就落伍啦!!!