R语言物种丰度矩阵的实现基本操作与生物物种多样性指数计算
Posted DU Group
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言物种丰度矩阵的实现基本操作与生物物种多样性指数计算相关的知识,希望对你有一定的参考价值。
本文由“壹伴编辑器”提供技术支持
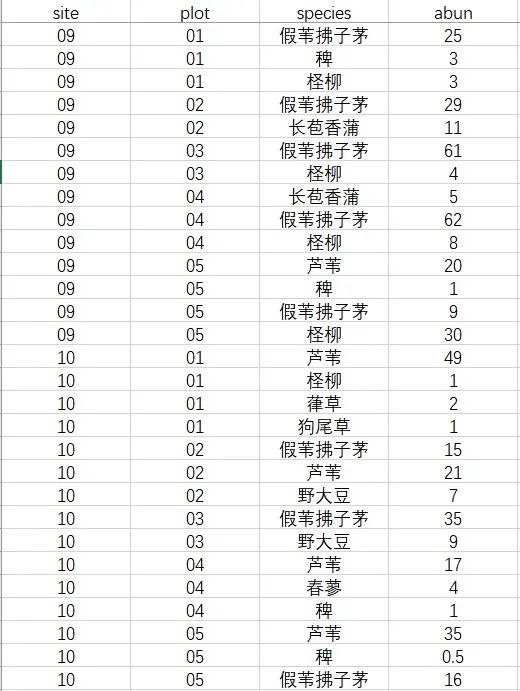
图1 植物群落数据举例
代码如下:
#读取数据
rawdata= read.csv("rawdata.csv", header = T) #第一行为表头
library(doBy)#加载R包(需提前安装)
rawdata.site= summaryBy(abun ~ site + species, data=rawdata, FUN=mean)
#将abun列的数据按照每个样地中的
#每个物种为单位(site +species)
#计算均值
names(rawdata.site)[3]="abun"
#对均值列的列名重新命名为“abun”
head(rawdata.site)#查看前六行的数据
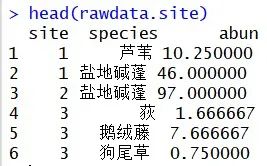
代码如下:
tram.sitedata= function(data1)
{Species=unique(data1$species)
#提取所有的不重复物种名称并赋给
#Species向量
Site=unique(data1$site)
#提取所有不重复样方编号并赋给Site向量
n=length(Species)
#计算不重复物种数并赋值给n
m=length(Site)
#计算不重复的样方数并赋值给m
newdata=as.data.frame(matrix(0,m,n))
#构建一个新m×n的矩阵,然后将矩阵
#转为数据框,才能添加行名和列名
names(newdata)=Species
#将列名改为物种名称
row.names(newdata)=Site
#将行名改为样地的名称
#做一个行循环,然后进行填空
for(iin 1:m)
{newdata1=data1[data1$site==Site[i],]
#提取原始数据第i个样地的数据
newspecies=newdata1$species
#提取第i个样地的物种名
newn=length(newspecies)
#提取第i个样地的物种数
#做一个列循环,看多少个物种,
#就插入多少数值
for (jin 1:newn)
{Ncol=which(Species==newspecies[j])
#提取第i个样地的物种所在的位置
newdata[i,Ncol]=newdata1$abun[j]
#第i行物种所在的位置依次#填入
#当前的物种多度值
}
}
return(newdata)#返回结果
}
spe.matrix= tram.sitedata(rawdata.site)#丰度矩阵
library(vegan)#(需提前安装)
spe.rel.matrix<- decostand(spe.matrix, "total")#相对丰度矩阵
write.csv(spe.rel.matrix,"spe.rel.csv")
#输出结果
将数据读出到spe.rel.csv文件中,结果如下:
图2转换后的相对丰度矩阵
根据物种相对丰度矩阵,我们可以计算生物多样性指数(以物种多样性为例)。
代码如下:
library(vegan)
Richness=rowSums(spe.rel.matrix>0)#物种丰富度指数
Shannon=diversity(spe.rel.matrix,"shannon")
#shannon多样性指数
Simpson=diversity(spe.rel.matrix,"simpson")
#Simpson多样性指数
H=diversity(spe.rel.matrix)
Pielou=H/log(Richness) #Pielou均匀度指数
完
本文编辑
2016级博士 衣世杰
以上是关于R语言物种丰度矩阵的实现基本操作与生物物种多样性指数计算的主要内容,如果未能解决你的问题,请参考以下文章