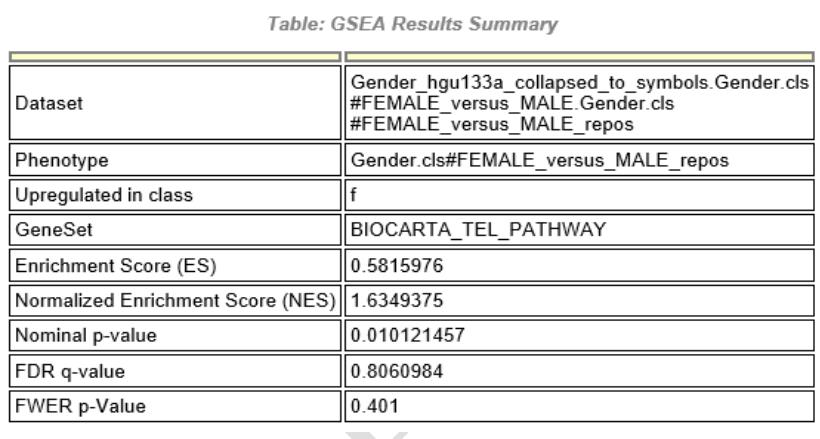
GSEA分析流程
Posted 修罗神天道
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GSEA分析流程相关的知识,希望对你有一定的参考价值。
1. 简介
GSEA 是 2003 年提出来的一种对表达谱芯片进行分析的方法,并被编制成软件。它的主要目的就是确定预先定义的基
因集(具有相同或相似的功能,或位于同一染色体相邻位点的一群基因)在表达谱芯片结果中是否有显著性。
GSEA 分析过程分为 5 步:
1. 基因知识库的获得;
2. 根据基因表达谱数据对所有基因进行排序;
3. 计算富集得分(enrichment score,ES);
4. 估计显著性水平;
5. 进行多重假设检验。
GSEA 能够鉴定疾病发生过程中潜在的及起决定作用的遗传改变或信号转导通路,从而把表达谱芯片数据和生物学意
义偶联起来。一些研究者通过 GSEA 鉴定了糖尿病患者肌肉组织中氧化磷酸化信号通路的下调以及腺癌患者 K-RAS
的下调等
2. 下载安装(GSEA v3.0)
下载地址 http://software.broadinstitute.org/gsea/downloads.jsp (注册后可免费下载)
下载后安装打开(依托的 java 运行环境按照提示下载安装)
初始界面如下:
3. 数据准备
3.1 数据要求:
1. 可使用 Excel 或者某种文本编辑器创建或者编辑 GSEA 文件
2. 文件中以制表符(table)隔开
3. 输入文件名称不要包含连字符号(hypens, -)
支持的文件格式类型
1. Expression Data Formats(芯片表达谱数据集文件)
(1) GCT: Gene Cluster Text file format (*.gct)
(2) RES: ExpRESsion (with P and A calls) file format (*.res)
(3) PCL: Stanford cDNA file format (*.pcl)
(4) TXT: Text file format for expression dataset (*.txt)
2. Phenotype Data Formats(表型数据文件)
( 1) CLS: Categorical (e.g tumor vs normal) class file format (*.cls)
( 2) CLS: Continuous (e.g time-series or gene profile) file format (*.cls)
3. Gene Set Database Formats ( 功能基因集文件)
( 1) GMX: Gene MatriX file format (*.gmx)
( 2) GMT: Gene Matrix Transposed file format (*.gmt)
( 3) GRP: Gene set file format (*.grp)
( 4) XML: Molecular signature database file format (msigdb_*.xml)
4. Microarray Chip Annotation Formats ( 芯片注释文件)
( 1) CHIP: Chip file format (*.chip)
5. Ranked Gene Lists
( 1) RNK: Ranked list file format (*.rnk)
3.2 主要的文件格式简介:
1. GCT: Gene Cluster Text 文件格式 (*.gct) 芯片表达谱数据集文件
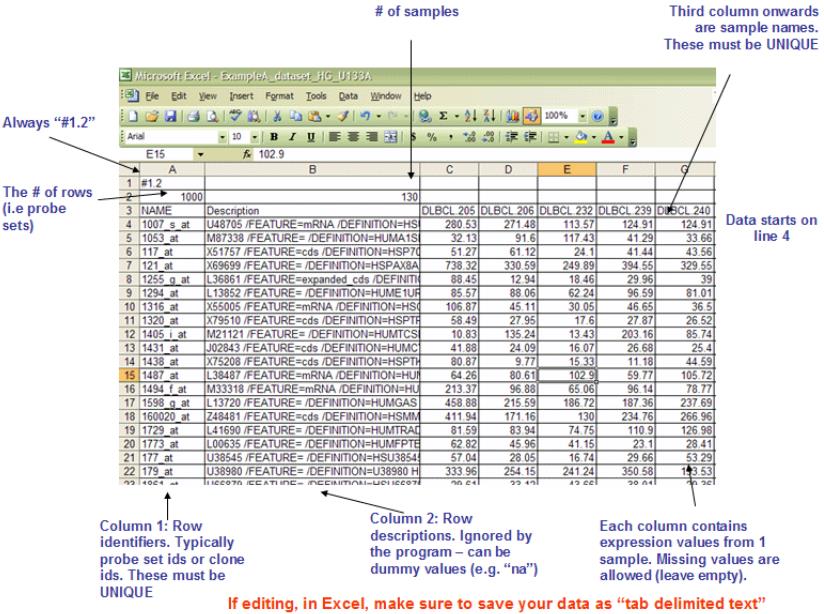
第一行:版本信息(一般是#1.2);
第二行:第二行为两个数值,第一个数值是基因数目,第二个数值是样本数目,中间以制表符( tab)隔开;
第三行:数据表头 NAME:基因名称, Description: 基因相关描述或注释,其他列:各样本名称,中间以制表
符( tab)隔开;
第四行及以后:数据内容为:基因名称,基因相关描述或注释和基因在各样本表达值数据,中间以制表符
( tab)隔开。
2. CLS: Categorical (e.g tumor vs normal) class file format (*.cls) 表型数据文件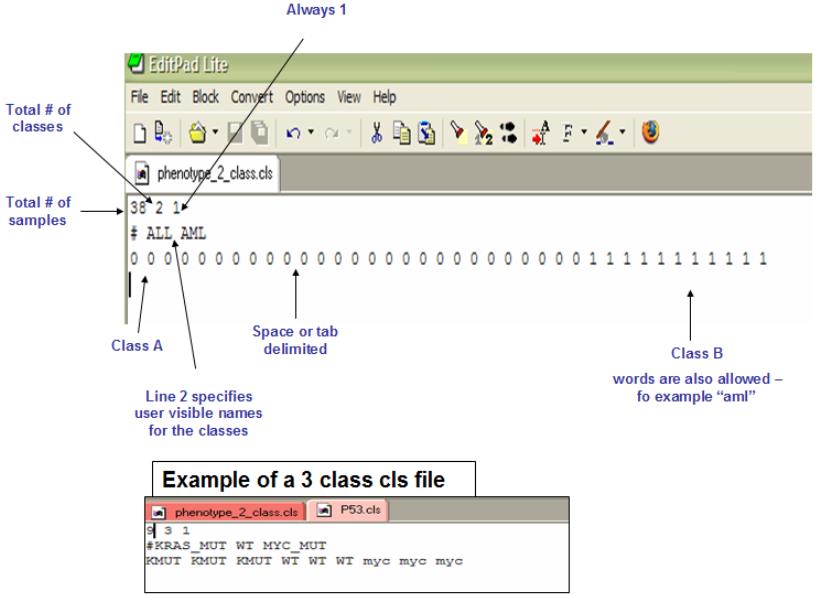
Cls 文件包含三行内容
第一行为样本数目,分类数目和 1, 中间以空格隔开,也可以 tab 隔开;
第二行为#, 类别一的名字, 类别二的名字……, 中间以空格隔开, 也可以 tab 隔开;
第三行为各样本的类别名,顺序为 gct 文件中样本顺序, 中间以空格隔开,也可以 tab 隔开。
前面为处理,后面为对照
3. 功能基因集文件
功能基因集文件即是位于 MSigDB 中的各个信息文件,可有.gmx 和.gmt 两种格式。当功能基因集文件较多
时,使用.gmt 格式更便于保存,但当功能基因集文件数<256 时,应用.gmx 格式更利于发挥 EXCEL 的编辑优势。通
常在 GSEA 网站可直接下载.gmt 格式的功能基因集文件使用,而无需自行创建。
4. 芯片注释文件
芯片注释文件格式为"*.chip",常用的芯片类型其注释文件都可在 GSEA 网站下载。
虽然此文件不直接在 GSEA 算法中使用,但它用于注释输出结果,也可用于将表达数据集中的每个探针集
合折叠为单个基因载体。
其他文档有相应的别的要求,如果文档格式出错,会有完整的提示,注意文档格式的要求很严格,稍有不对就无法
完整导入数据。各个文档格式的详细要求:
http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats
4. RUN
4.1 数据导入(点击左侧的 load data)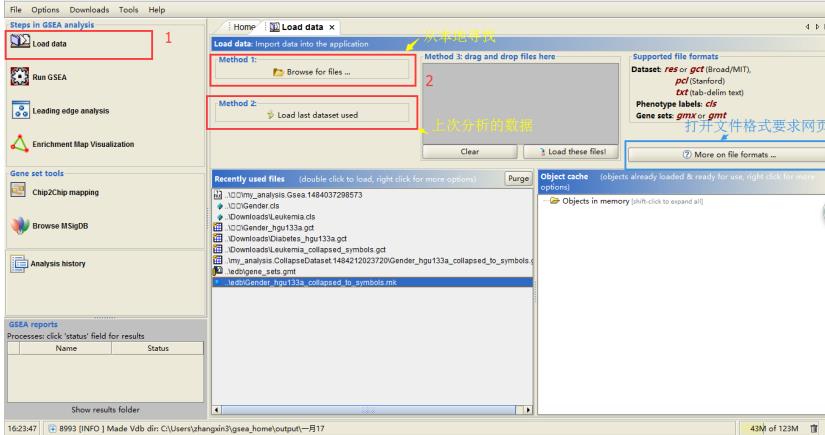
4.2 RUN 环节
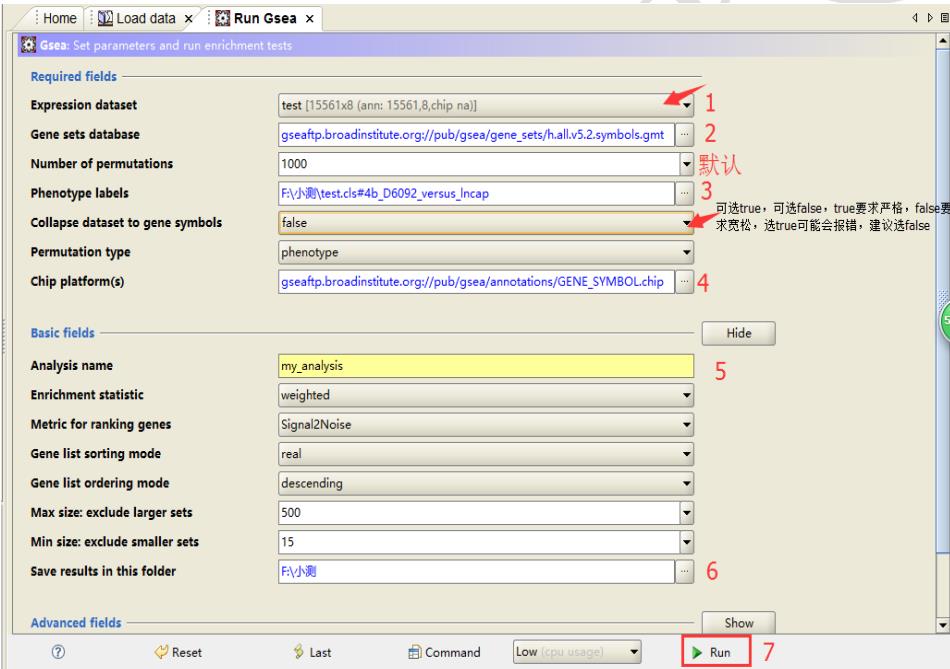
成功导入数据后,可以进入 run 环节,点击 Run GSEA 进入下图所示页面: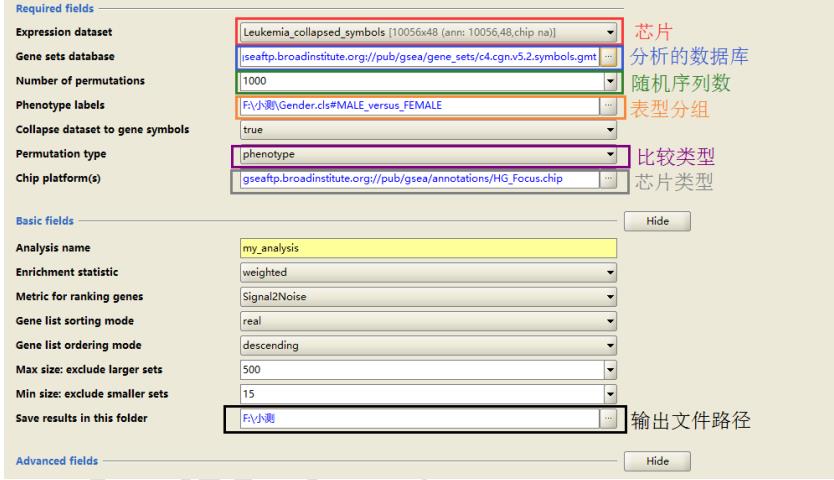
各个文件选择好后,可以命名分析名称,选择输出文件路径, cpu 使用情况等。一切就绪后,点击下方的 Run 按钮
即可开始运行。
注意: 1.输出路径默认是本地用户组用户文件目录下的 gsea_home/output/X 月 X 文件夹内(可更改)
2.cpu 使用情况有 Low、 high 和 Normal 三个选项。选择代表 cpu 的使用情况, 高、中、低三种情况。
运行具体过程可点击下图所示位置,可弹出相应应用信息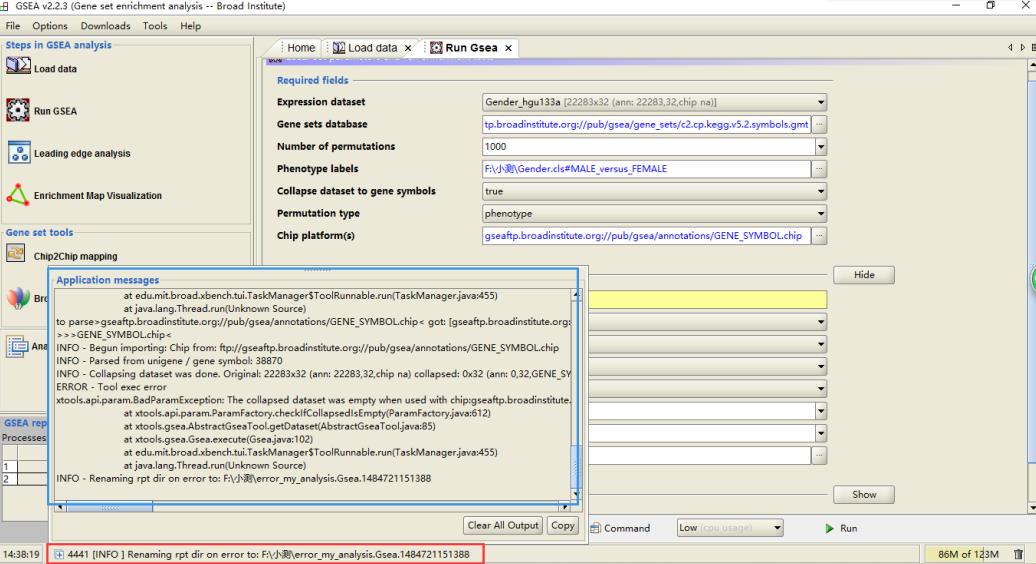
4.3 结果生成
观察左下角的 GSEA reports 框可观察结果产生情况,状态有三种: 1.Running (正在运行) 2.Error (出错) Success (成
功)。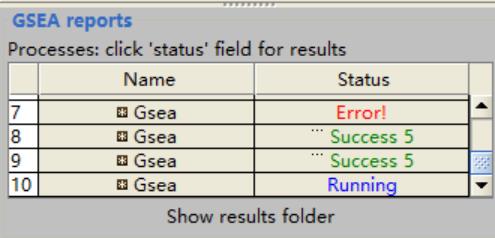
点击 Running 后可选择调整 cpu 使用情况(low、 normal 或者 high)和暂停正在运行的任务;
点击 Error 后会出现详细的报错信息,可以查看报错的详细信息或者帮助等;
点击 Success 后跳转到一个 GSEA 报告页面。具体情况如下文所述;
点击下方的 Show results folder 会跳转到默认输出目录中,如果更改了输出目录,需要自己手动寻找结果目录。
5. 结果解读
1) Error 也会在结果文件夹内产生一个 error 文件夹,里面存在两个文件, index.html 和 xtools.css。基本没什么用
处,可直接删除。
2) Success 会在结果文件夹中产生以 Analysis name.Gsea.数字代码命名的一个文件夹,里面有各种结果文件,直接
打开其中的 index.html 文件可打开结果报告网页。或者直接点击软件左下角, GSEA reports 框中的相应分析结
果后的 Success 也可直接跳转至结果报告页面。
结果报告例子如下图所示: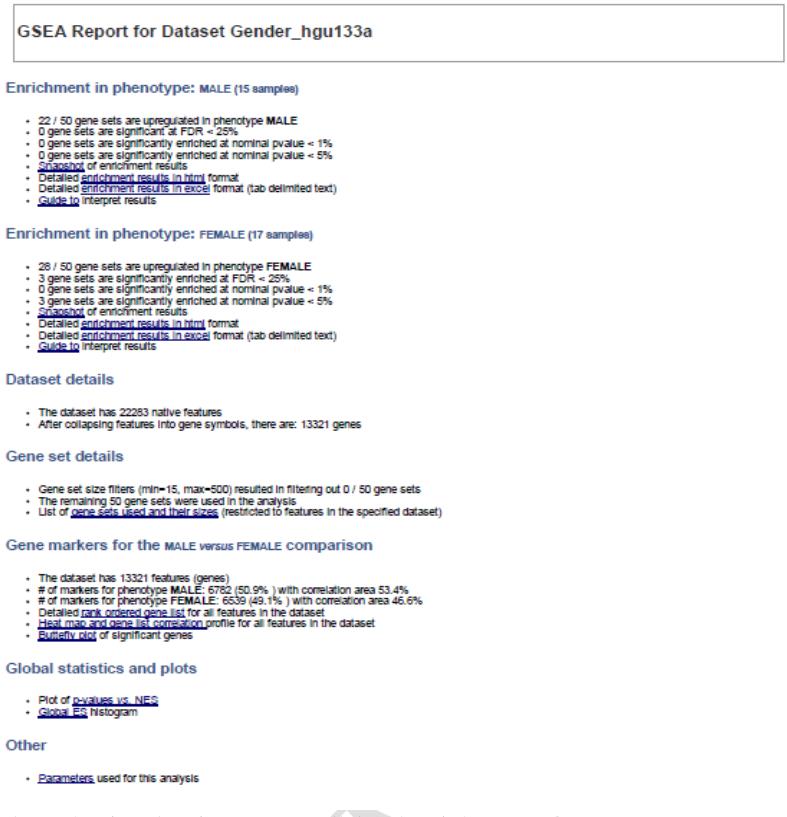
结果报告一般分为七个部分(cls 文件中包含两个类别)
(1) Enrichment in phenotype: class 1 (sample num)
类别 1 中,在研究的 m 个基因子集中,有 n 个基因子集的表达上调(n/m);
75%的正确率支持其中的 x 个基因子集中的基因表达上调;
有 y 条基因子集中的基因表达上调的名义 p 值小于 0.01;
有 z 条基因子集中的基因表达上调的名义 p 值小于 0.05。
(2) Enrichment in phenotype: class 2 (sample num)

类别 2 中,在研究的 m 个基因子集中,有 n 个基因子集的表达上调(n/m);
75%的正确率支持其中的 x 个基因子集中的基因表达上调;
有 y 条基因子集中的基因表达上调的名义 p 值小于 0.01;
有 z 条基因子集中的基因表达上调的名义 p 值小于 0.05。
(3) Dataset details
原始芯片数据包含探针数目以及这些探针代表的基因数目。
(4) Gene set details
在选择的数据库中,基因集中包含基因子集数目,在定义的选择标准下(功能基因子集中所含基因数大于 15小于 500),有多少基因子集被过滤,有多少基因子集用于分析。
(5) Gene markers for the class 1 versus class 2 comparison
(6) Global statistics and plots
(7) Other

分析所用的各个参数
3) 部分结果详解
点击结果报告中分组富集信息中的 Snapshot 超链接
其结果中有四个关键的统计量值,分别是富集得分(enrichment score,ES),标准化富集得分(normalized enrichmentscore, NES),错误发现率(false discovery rate, FDR)和名义 P 值(nominal P value)。
富集得分(enrichment score,ES)
ES 是 GSEA 分析的原始结果, ES 反映 S 在秩列表 L 顶部过表达的程度。 设总基因个数为 N, L=[g1,g2,……gN] 是依据各基因与表型间相关性 r 排序的基因列表。 基因集 S 富集得分 ES(S)的计算基于基因列表 L,从 ES(S)=0 开始,遇
到 S 中的基因时增加 ES(S),相反则减小,增加或减少的幅度依赖于基因与表型的相关性 r(𝑔𝑗) =𝑟𝑗、 基因集的大小M、总基因个数 N。
根据基因列表 L,从 i=1 到 N,若 i∈S,则 ES(S)增加|𝑟𝑖|𝑝/ ∑𝑔∈𝑆 |𝑟𝑖|𝑝; 若 i∉S,则 ES(S)减小 1/N-M。 当 ES 值为正,表示某一功能基因集富集在排序序列的前方,当 ES 值为负,表示某一功能基因集富集在排序序列的后方。 富集得
分取绝对值的最大的数值。
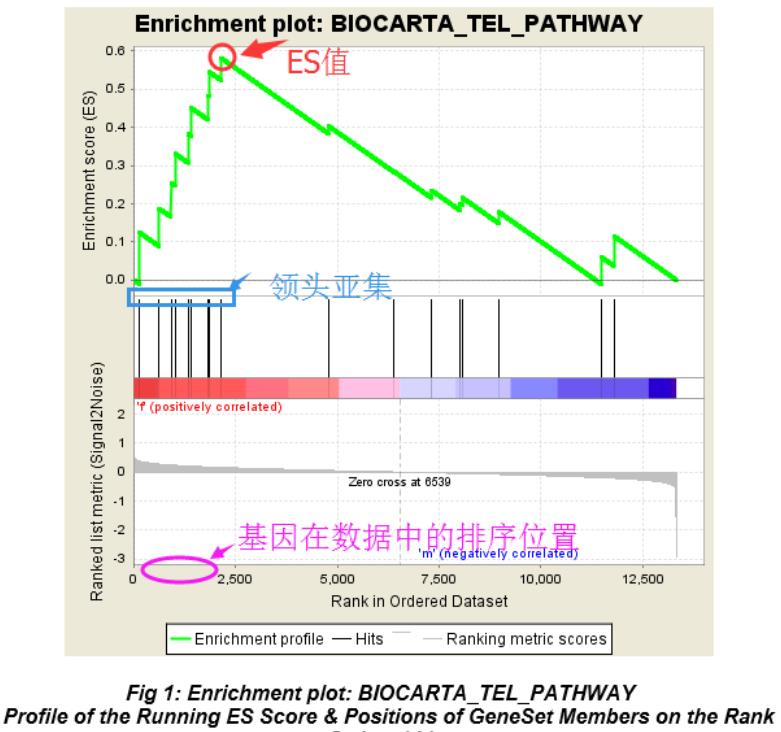
图上部的曲线表示动态 ES 值,最高点表示此通路的 ES 值。图的中间部分表示杂交数据的排序序列, 黑色竖线表示在此通路中出现的基因。此外还有一个领头亚集(leading edge subset),领头亚集中的基因是指对 ES 值贡献最大的
基因集合。当 ES 为正值时,领头亚集位于 ES 值对应排序序列之前,反之,则位于 ES 值对应排序序列之后。显然,领头亚集的出现说明一方面这些基因在通路中有富集,非散在分布,另一方面,说明这些基因在通路中有共同的表
达趋势。显然,在 ES 图中出现领头亚集的形状的,表明这个功能基因集在定义的实验条件下具有更显著的生物学意义。此外,图下部曲线表示的是排序值。排序值沿芯片数据的排序序列由大到小分布,在分类表型数据文件,正
值表示该基因与第一个表型相关,负值表示与第二个表型相关。在连续表型数据文件,如时间系列,正值表示相关而负值表示负相关或没有相关性。 在所有的基因中,如果出现一个基因属于这个组合并且表达量在对应组里面表达
高于另一组,富集分数就增加,反之就下降。
标准化富集得分(normalized enrichment score, NES)
由于 ES 是根据分析的芯片数据集中的基因是否在一个功能基因集中出现来计算的,但各个功能基因集中所包含的基因数不同,而且不同的功能基因集与芯片数据间的相关性也不同,因此在比较芯片数据集在不同功能基因集中的
富集程度时,需要对 ES 进行标准化处理。 GSEA 定义 NES 为: NES = (某一功能基因集的 ES)/(数据文件集所有随机组合得到的 ES 的平均值)NES 是功能基因集富集分析结果的主要统计量。NES 是建立在数据文件集所有随机组合得到的 ES 平均值的基础上,因此,数据随机组合方法,随机组合次数,或表达数据文件集大小的改变都会影响 NES。
错误发现率(false discovery rate, FDR)
FDR 描述的是一个估计的可能性,即当一个功能基因集的 NES 值确定后,判断其中可能包含的错误的阳性发现率。
例如, FDR=25%意味着对此 NES 值的确定, 4 次中可能有 1 次是错误的。在 GSEA 的结果报告中,高亮显示了 FDR值小于 25%的富集功能基因集,因为从这些富集功能基因集中最可能产生有意义的科学假设及促进进一步深入研究。
在大多数情况下,选择 FDR 值为 25%来判定是否是富集的功能基因集是合适的,因为通常用于分析的芯片表达数据之间,大部分都缺乏一致性,而且每次分析的功能基因集数目不多。但是,当分析的芯片数据集较小,分析时选择
的是探针间的随机组合(gene-set permutation)而不是表型间的随机组合(phenotype permutation), P 值采用的严格度又不高时,应该选择更加严格的 FDR 界值,如 FDR=5%。一般而言, NES 的绝对值越大, FDR 的值就越小, 说
明富集度越高的功能基因集,分析结果的可信度就越高。
名义 P 值(nominal P value)
名义 P 值描述的是针对某一个功能基因子集得到的富集得分的统计显著性,很显然, P 值越小说明基因的富集性越好。但在 GSEA 分析结果的四个参数中,只有 FDR 值进行了功能基因子集大小和多重假设检验的校正,而 P 值没
有。因此,当分析结果中出现一个高度富集的功能基因子集,具有很小的名义 P 值但却有很大的 FDR 值时,往往意味着其实和其它功能基因子集相比较,它的富集并不是很显著。原因可能是用于分析的芯片数据样本量较少,或
者是杂交信号微弱,又或者是选择的功能基因子集并没有很好地反映样本的生物学意义。在 GSEA 的报告中, P=0.0 意味着实际的 P 值小于 1/ 随机组合的次数。例如,当选择的随机组合数为 100 时,报
告中的 P=0.0 即是说实际的 P 值小于 0.001。所以提高随机组合数,就能得到更精确的 P 值。一般运行 GSEA 时随机组合次数都选择 1000,但不要超过 1000,因为一旦超过 1000,运行 GSEA 程序可能会耗尽内存。
结果详解(http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html?_Interpreting_GSEA_Results )
6. 实例
6.1 转录组项目
1. 数据准备
(1) 各样本表达量文件
(2) 各样本的分类情况
(3) *.gct 文件的构建
a) AllSamples.GeneExpression.FPKM.xls 文件,其中包含各个样本的表达量值。 其中包括:
gene_id、transcript_id(s)、sample1_FPKM、sample2_FPKM、sample4_FPKM… Symbol 、Cellular Component、
Molecular Function 、 Biological Process、 Kegg Orthology、 Nr Description
b) 从中获取*.gct 文件需求的各样品的表达量(sample_FPKM) 以及基因名(symbol), 可获取相关注释描述(如 Nr Description)。
例如:
提取前:
| gene_id | transcript_id(s) | sample1_FPKM | sample2_FPKM | … | Symbol | … | Description |
| XXX | XXX | XXX | XXX | XXX | XXX | XXX | XXX/NA |
提取后:(注意中间为制表符或者空格)
| Symbol | Description | sample1_FPKM | sample2_FPKM | … |
| XXX | XXX/NA | XXX | XXX | XXX |
c) 根据 gct 文件格式的要求,需要统计基因数目和样本数目(注意中间为制表符或者空格)
| #1.2 (版本信息一般是#1.2) | ||||
| Num(基因总数) | Num(样本总数) | |||
| Symbol | Description | sample1 | sample2 | … |
| XXX | XXX/NA | XXX | XXX | XXX |
(4) *.cls
根据 cls 文件格式的要求配置文件
例如:
说明 a、 中间以制表符或者空格隔开;
b、第一行依次表示样本数、类别数、 1;
c、第二行依次表示#、类别 1 的名字、类别 2 的名字;
d、第三行为 sample1 的类别、 sample2 的类别、 sample3 的类别(注意顺序和 gct 文件中样本顺序一致。
2. 运行
(1) Load data
(2) 进入 RUN 阶段
3. 结果
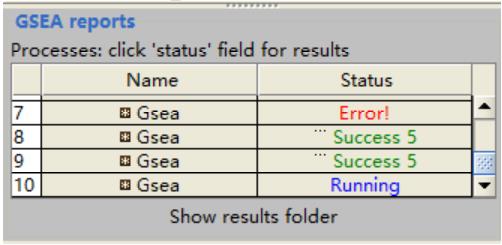
左下角的 GSEA reports 中出现 Success 5 时意味成功,点击 success 5 会出现结果的网络版报告。出现 Error!意味出错,点击 Error!可展示详细报错内容,根据报错信息,调整后跑出成功结果。
最终结果保存在 RUN 步骤中第 6 步选择的 Save results in this folder 的文件夹中。
6.2 RNA-Seq
类似转录组重测序,从GeneExp 提取 all.gene.FPKM.xls
文件,后构建 gct 文件和 cls 文件。后续步骤类比上述。
6.3 注意事项
1. 建议使用 symbol,根据 chip platform 选择 Expression dataset(*.gct)。
2. Gene sets database 请根据需求选择。
3. Collapse dataset to gene symbol
如果折叠数据集(将数据集折叠到基因符号参数= True),则折叠数据集中的基因通过基因符号识别,因此基因集中的基因标识符必须是基因符号;
如果不折叠数据集(将数据集折叠到基因符号参数= False),则基因集中的基因标识符必须与表达式数据集中的基因标识符相同。
4. 左下角的 GSEA reports 中出现 Error! 后请点击 Error! 后查看详细的报错信息和由软件提供的解决方案,更改错误后重新运行!
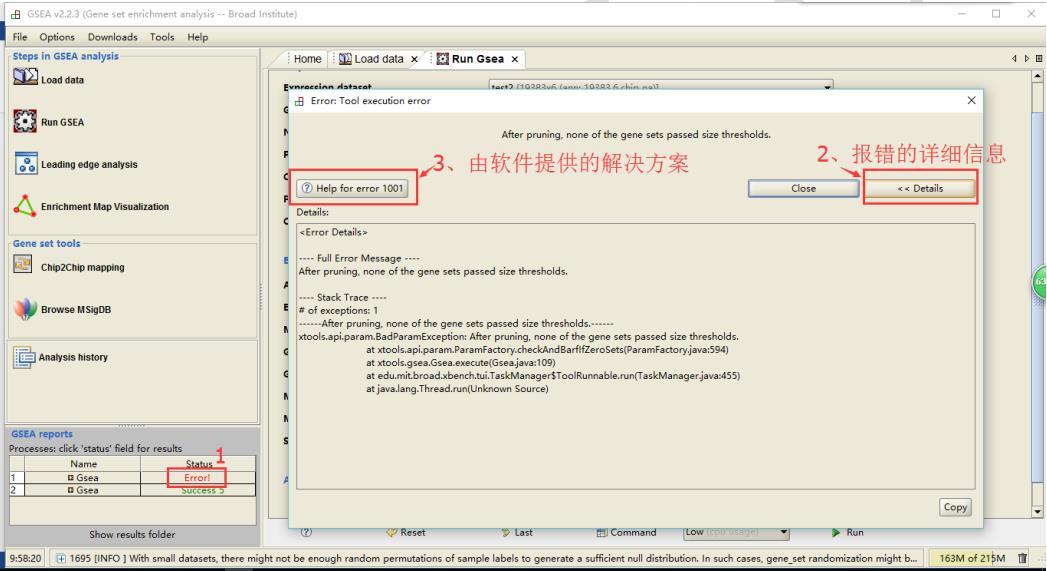
5.根据样本数目选择对应的 Metric for ranking genes
7. 附录
7.1 Molecular Signatures Database v6.1(http://software.broadinstitute.org/gsea/msigdb/index.jsp)
分子标记数据库(MSigDB)中的 17786 基因集分为 8 个主要集合和几个子集。
1) H: hallmark gene sets(browse 50 gene sets)
Hallmark 基因集合总结和代表特定的明确定义的生物状态或过程,并显示相关的表达。这些基因组基于鉴定基因组重叠和保留显示坐标表达的基因的计算方法产生。 Hallmark 减少噪声和冗余,并为 GSEA 提供更好的划定的生物空间。
To cite your use of the collection, and for further information, please refer to Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell
Syst. 2015 Dec 23;1(6):417-425.
2) C1: positional gene sets (browse 326 gene sets)
对应于每个人染色体和具有至少一个基因的每个细胞遗传带的基因集。 (细胞遗传位置从 HUGO 和 Unigene解析。当存在冲突时,使用 Unigene 条目。)这些基因组有助于鉴定与染色体缺失或扩增,剂量补偿,表观遗传沉默, 和其他区域效应。
3) C2: curated gene sets (browse 4738 gene sets)
此集合中的基因集来自以下方面:
在线途径数据库:代表代谢和信号通路的基因集从此处列出的在线途径数据库导入。
生物医学文献:在过去几年中,微阵列研究已经鉴定了几种重要的生物和临床状态(例如癌症转移,干细胞特征,耐药性)的标志。许多具有这些标志的基因集,最初是作为表格出版在论文中的,可提取出来作为 GSEA 分析的基因集。因此,其编辑了具有公开的基因表达标志的微阵列文章的列表,并且从每篇文章中,从主文本或补充信息的表中提取一个或多个基因组。目前,该集合包括来自超过 340 篇 PubMed 文章的基因集。其正在努力创建一个更自动化的方法来从文献中制定基因集。
L2L:从公开的哺乳动物微阵列研究中汇编的基因集MYC 靶基因数据库:来自约翰霍普金斯大学医学院 MYC 靶基因数据库的 Chi Dang 博士策划的基因组。
( a) CGP: chemical and genetic perturbations(browse 3409 gene sets)
代表遗传和化学变化表达标志的基因集。 许多这些基因集成对存在,例如: xxx_UP( xxx_DN)基因集代表由变化诱导(抑制)的基因。 每个基因集的基因组页面列出了它所基于的 PubMed 引文。
( b) CP: Canonical pathways(browse 1329 gene sets)
来自通路数据库的基因集。 通常,这些基因集是由领域专家编译的生物过程的规范表示。
( c) CP:BIOCARTA: BioCarta gene sets(browse 217 gene sets)
来自 BioCarta 途径数据库的基因集 http://www.genecarta.com/
( d) CP:KEGG: KEGG gene sets(browse 186 gene sets)
来自 KEGG 途径数据库的基因集 http://www.genome.jp/kegg/pathway.html
( e) CP:REACTOME: Reactome gene sets(browse 674 gene sets)
来自 Reactome 途径数据库的基因集 http://www.reactome.org/
4) C3: motif gene sets(browse 836 gene sets)
包含共享在人,小鼠,大鼠和狗基因组中保守的顺式调节元件的基因集。 这些基序被编目,同时这些基序表示启动子和 3'-UTR 中的已知或可能的调节元件。 这些基因组使得可能与微阵列实验中的变化与保守的推定的顺式调节元件相联系。
( a) MIR: microRNA targets(browse 221 gene sets)
含有共享 3'-UTR 微小 RNA 结合基序的基因的基因集
( b) TFT: transcription factor targets(browse 615 gene sets)
含有共享在 TRANSFAC (version 7.4, http://www.gene-regulation.com/ )数据库中定义的转录因子结合位点的基因的基因集。 这些基因组中的每一个由 TRANSFAC 记录注释。
5) C4: computational gene sets(browse 858 gene sets)
计算基因集是通过挖掘癌症导向微阵列数据结果的集合。
( a) CGN: cancer gene neighborhoods(browse 427 gene sets)
由以 380 个癌症相关基因为中心的表达邻域定义的基因集合(Brentani, Caballero et al. 2003)。
( b) CM: cancer modules(browse 431 gene sets)
由 Segal 等人定义的基因集合(Segal et al. 2004)。 作者从各种资源(如 KEGG, GO 等)编辑了基因集( '模块')。 通过挖掘癌症相关微阵列数据的大纲,他们确定了 456 个这样的模块,在各种癌症条件下显著变化。
6) C5: GO gene sets(browse 5917 gene sets)
该集合中的基因集是从 Gene Ontology 项目( www.geneontology.org )的中导出的。基因集是基于 GO 术语( go-basic.obo, 2016 年 5 月 3 日下载)及其与人类基因的相关性的( gene2go,从 2016 年 5 月 3 日从 NCBIFTP 服务器下载)。分为三部分:分子功能( MF),细胞组分(CC)和生物过程( BP)。基因产物可能与一个或多个分子功能相关或位于其中。
GO 注释由描述特定 GO 项和基因产物之间的关联所基于的工作或分析的特定参考相关联的 GO 项组成。每个注释还必须包括证据代码,以指示如何支持对特定术语的注释( http://geneontology.org/page/guide-goevidence-codes )。GO 基因集省略了非常广泛的类别,如生物过程,同时也省略了具有少于 10 个基因的 GO 集(NCBI Entrez Gene ID)。如果 Jaccard 的系数大于 0.85,其将集合定义为“高度相似”。对于每一对高度相似的集合,其保持最大的集合并重复该过程,直到所有这样的对被解决。
(a) BP: GO biological process(browse 4436 gene sets)
生物过程(BP) http://www.geneontology.org/GO.process.guidelines.shtml
(b) CC: GO cellular component(browse 580 gene sets)
细胞组分(CC) http://www.geneontology.org/GO.component.guidelines.shtml
(c) MF: GO molecular function(browse 901 gene sets)
分子功能(MF) http://www.geneontology.org/GO.function.guidelines.shtml
7) C6: oncogenic signatures(browse 189 gene sets)
该基因集代表在癌症中经常失调的细胞途径。 大多数是直接从 NCBI GEO 的微阵列数据或者涉及已知癌基因的变化的内部未公开的分析实验产生的。 此外,少数致癌基因是从科学出版物找出的。
8) C7: immunologic signatures(browse 4872 gene sets)
免疫学标记集合(也称为 ImmuneSigDB)由代表细胞类型,状态和免疫系统内的变化的基因组组成。从人和人免疫学中已发表的研究中找寻的。其首先捕获在免疫学文献中公布的相关微阵列数据集的具有保存到基因表达综合征(GEO)的原始数据。 对
于每个公开的研究,鉴定相关比较(例如 WT 对 KO;治疗前和治疗后等),并且创建简短的,生物学上有意义的描述。以相同的方式处理和归一化所有数据后用来鉴定基因组。
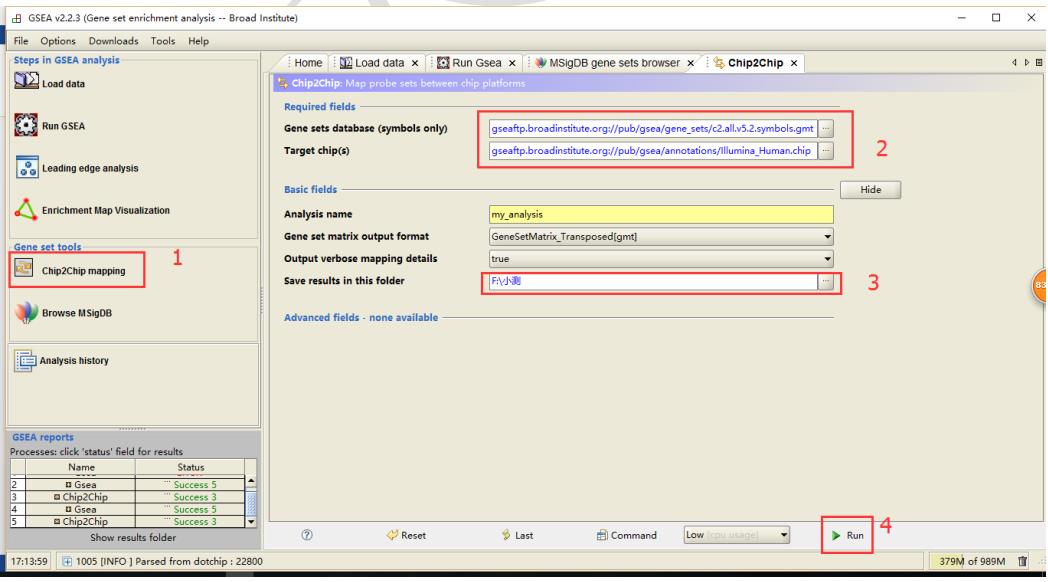
7.2 Chip2Chip mapping
GSEA 软件提供 Gene sets database 与 target chip 比对的功能,最后结果为两者的交集,最后产生的结果为两者交集的 gmt 文件,可用于 GSEA 分析。
8. FAQ
8.1 Error: Tool execution error
运行开始后即跳出如下错误: Connection reset by peer: socket write error.
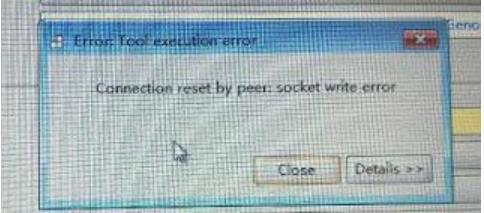
A:此类错误一般由网络连接问题导致。 由于选择了在线的 GSEA 基因集文档和 chip 文档,因此软件在运行时需要连接相关在美国的 ftp 服务器,这可能会被一些学校、公司的网络所禁止。 为了避免在线分析, 我们只需要从官网页面 http://software.broadinstitute.org/gsea/downloads.jsp 下载我们所需要的 gmt、 chip 文件, 在 run 之前选择基因集等数据库时,选择 local 文件即可。
8.2 GSEA 支持什么芯片平台和物种?
A: 只要所需要分析的表达量数据有基因标识符(gene identifier),且能对应到 GSEA 的基因集中(gene sets), GSEA就能对此表达量数据做分析。GSEA 所利用的 MSigDB 中的基因都含有人类基因标识(human gene symbols), 其通过特殊的 chip file 将其他基因的标识转化为人类基因标识。 因此,需要 chip files 提供相关的 mapping 关系数据。所以,如果我们分析的数据是非人样本,我们需要确定是否利用 MSigDB 来进行分析。 具体情况如下:
1) 利用非人物种作为模式物种来研究人类。 MSigDB 是正确的选择来进行分析,只需提供合适的 chip file 即可;
2) 非人物种是研究的主要对象,没有需要去联合研究人类。MSigDB依然是可以选择的,只需要提供合适的chip file;
3) 非人物种是研究的主要对象, 且不希望利用 MSigDB。 客户需要自己提供 gene sets 文件(GMT 或 GMX 格式),
同样需要确认在 gene sets 中的基因标识能和测序结果的基因标识匹配得上, 如果不能匹配,则需要提供合适的 chip files 来进行匹配。
PS: GSEA 中自带的 chip files 都只提供了匹配到人类基因标识的关系,不支持匹配到其他物种。
8.3 RNA-Seq 数据和 Ensembl CHIP files
A: GSEA 利用基因表达数据文件中的 gene identifiers 跟 MSigDB 的 HUGO human gene symbols 比对, 比对关系则依赖于 chip files 的信息。 但在 RNA-seq 中, 针对参考基因组来源于 Ensembl 数据库的分析结果, GSEA 提供了相关 chip
file, 将人和鼠的 Ensembl IDs 转化为 HUGO human gene symbols。 目前支持的 Ensembl id 前缀是:人(ENSG 和 ENST),鼠(ENSMUSG 和 ENSMUST)。
在设置参数的过程中,一般建议 gene sets 利用 Hallmarks collection 做分析,对于选择哪种 chip file, 规则如下:
ENSEMBL_human_gene.chip => Ensembl ID prefix ENSG
ENSEMBL_human_transcript.chip => Ensembl ID prefix ENST
ENSEMBL_mouse_gene.chip => Ensembl ID prefix ENSMUSG
ENSEMBL_mouse_transcript.chip => Ensembl ID prefix ENSMUST
以上是关于GSEA分析流程的主要内容,如果未能解决你的问题,请参考以下文章