基因集富集分析 --- GSEA
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了基因集富集分析 --- GSEA相关的知识,希望对你有一定的参考价值。
参考技术A 基因集富集分析(Gene set enrichment analysis, GSEA)方法于2005年首次提出,题为Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles的相关论文发表于杂志PNAS,至今引用量已接近2万。
首先来看官网 https://www.gsea-msigdb.org/gsea/index.jsp 定义:
GSEA需要使用来自两个类别(标记为A或B)的样本的基因组表达谱进行分析。这些基因可以根据它们在两类间的差异表达,在一个秩次列表L中进行排序,给定一个先验定义的基因集S,GSEA的目标是确定S的成员是随机分布在整个L中,还是主要分布在顶部或底部。如基因集与表型相关则倾向于显示后一种分布。
GSEA方法有三个关键要素:
首先是 Enrichment Score (ES) 的计算 ,我们设表达谱数据D包含N个基因和k个样本,仍然是上述L和S,表型C,N个基因里包含在基因集S中的基因数N H ,控制步长权重的指数 p 。
全网太多教程,珠玉在前,没必要再赘述。
最后分享2个链接,感觉各种细节都讲得很好
https://www.bioinfo-scrounger.com/archives/561/
http://rvdsd.top/2018/04/04/BIoTools/GSEA%E7%AC%94%E8%AE%B0/
GSEA富集分析
GSEA定义
Gene Set Enrichment Analysis (基因集富集分析)用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一是已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义),一是表达矩阵,软件会对基因根据其于表型的关联度(可以理解为表达值的变化)从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。
(The gene sets are defined based on prior biological knowledge, e.g., published information about biochemical pathways or coexpression in previous experiments. The goal of GSEA is to determine whether members of a gene set S tend to occur toward the top (or bottom) of the list L, in which case the gene set is correlated with the phenotypic class distinction.)
这与之前讲述的GO富集分析不同。GO富集分析是先筛选差异基因,再判断差异基因在哪些注释的通路存在富集;这涉及到阈值的设定,存在一定主观性并且只能用于表达变化较大的基因,即我们定义的显著差异基因。而GSEA则不局限于差异基因,从基因集的富集角度出发,理论上更容易囊括细微但协调性的变化对生物通路的影响。
GSEA原理
给定一个排序的基因表L和一个预先定义的基因集S (比如编码某个代谢通路的产物的基因, 基因组上物理位置相近的基因,或同一GO注释下的基因),GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要聚集在L的顶部或底部。这些基因排序的依据是其在不同表型状态下的表达差异,若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。
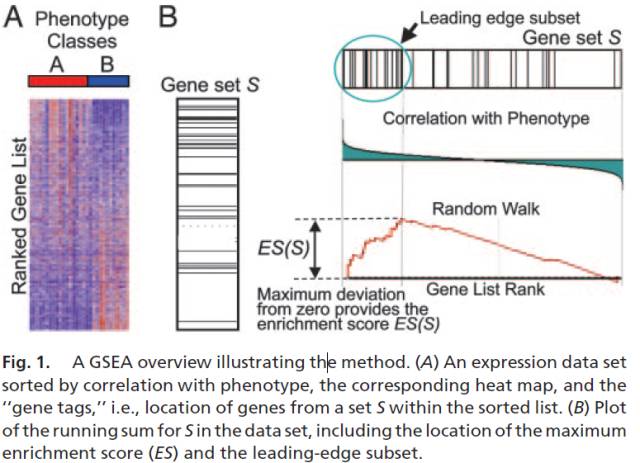
GSEA计算中几个关键概念:
-
计算富集得分 (ES, enrichment score). ES反应基因集成员
s在排序列表L的两端富集的程度。计算方式是,从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在s里面的基因,则增加统计值。遇到一个不在s里面的基因,则降低统计值。每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度)是相关的。富集得分ES最后定义为最大的峰值。正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。 -
评估富集得分(ES)的显著性。通过基于表型而不改变基因之间关系的排列检验 (permutation test)计算观察到的富集得分(ES)出现的可能性。若样品量少,也可基于基因集做排列检验 (permutation test),计算p-value。
-
多重假设检验矫正。首先对每个基因子集
s计算得到的ES根据基因集的大小进行标准化得到Normalized Enrichment Score (NES)。随后针对NES计算假阳性率。(计算NES也有另外一种方法,是计算出的ES除以排列检验得到的所有ES的平均值) -
Leading-edge subset,对富集得分贡献最大的基因成员。
GSEA分析
软件和基因集下载
(http://software.broadinstitute.org/gsea/downloads.jsp)
输入数据准备
-
表达矩阵。常见表达矩阵格式,
tab键分割,txt格式,第一列为基因名字(名字与注释数据库一致,同为GeneSymbol或EntrezID或其它自定义名字),第一行为标题行,含样品信息。也可为gct文件,具体见 http://blog.genesino.com/2014/08/GSEA-usages/
-
样品分组信息
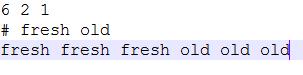
分组信息示例
-
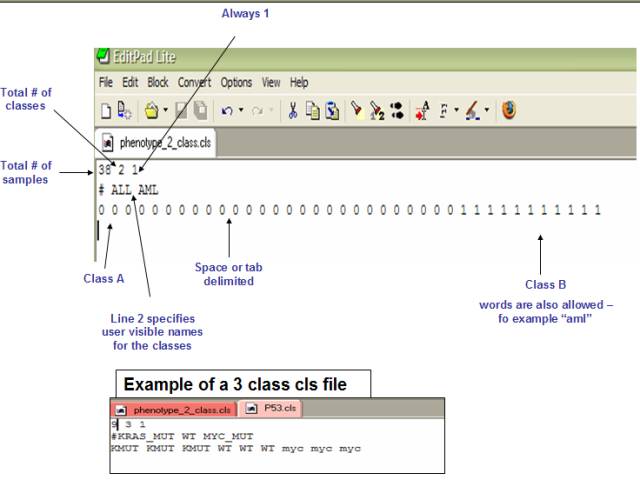
基因集信息

基因集信息示例
软件运行 (每一步的步骤如有不明确的参考文后第一个链接)
-
导入数据
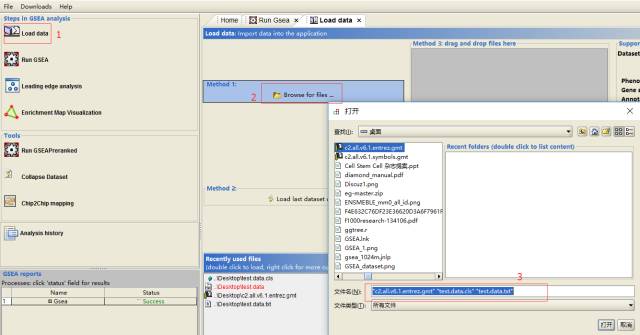
-
运行GSEA (若每组样品都有多于7个样品,则Permutation type选择
phenotype,结果理论上更好;否则选择gene_set)
-
设置好参数后,点击正下方的
run,等待运行结束,左侧出现success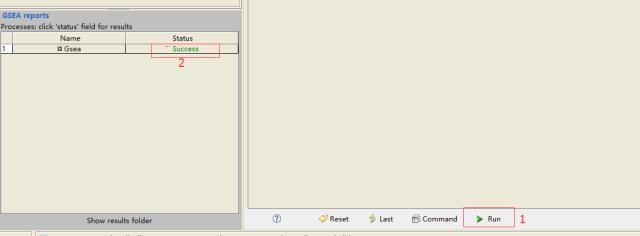
-
点击
success,查看结果
顺着网页的导航一步步去查看结果,有耐心就好。主要解释下,最常见的这种图。
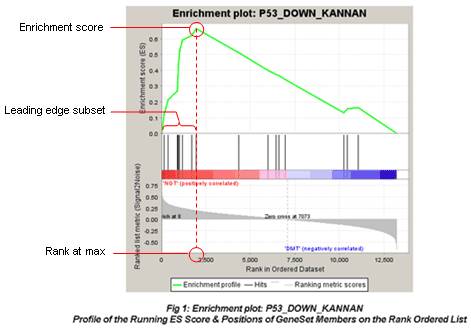
-
图最上面部分展示的是ES的值计算过程,从左至右每到一个基因,计算出一个ES值,连成线。最高峰为富集得分(ES)。在最左侧或最右侧有一个特别明显的峰的基因集通常是感兴趣的基因集。
-
图中间部分每一条先代表基因集中的一个基因,及其在基因列表中的排序位置。
-
最下面部分展示的是基因与表型关联的矩阵,红色为与第一个表型(
MUT)正相关,在MUT中表达高,蓝色与第二个表型(WT)正相关,在WT中表达高。 -
Leading-edge subset对富集得分贡献最大的基因成员。若富集得分为正值,则是峰左侧的基因;若富集得分为负值,则是峰右侧的基因。 -
FDR GSEA默认提供所有的分析结果,并且设定FDR<0.25为可信的富集,最可能获得有功能研究价值的结果。但如果样品数目少,而且选择了gene_set作为Permumation type则需要使用更为严格的标准,比如FDR<0.05。
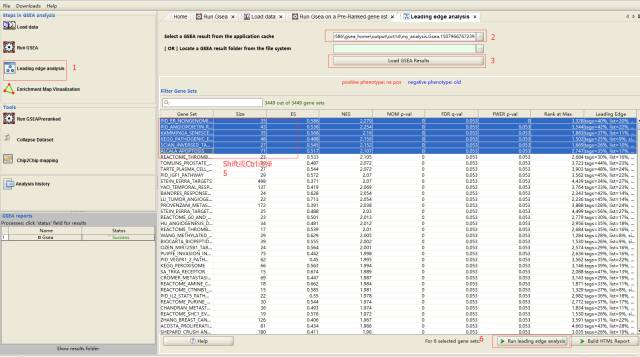
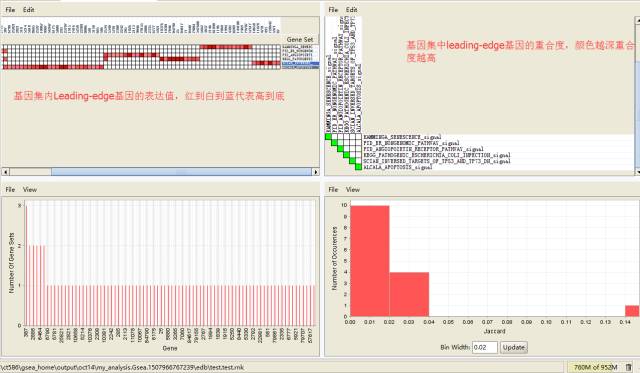
Leading-edge分析
主要对筛选感兴趣的基因有意义;选择一个或多个显著富集的基因集,查看其内Leading-edge基因的表达和重叠状态。
MSigDB
GSEA团队整理好的基因集,可用于注释,也可下载下来搜寻自己感兴趣的方向的基因作为一个补充。每个注释都提供了基于Gene Symbol和Entrez ID的索引表格。

参考
-
较早记录的一篇GSEA的使用,有脚本可以转换表达矩阵为
gct,cls文件作为GSEA的输入。文档为英文,但软件操作步骤还算详细,可配合着看。http://blog.genesino.com/2014/08/GSEA-usages/ -
最开始学习的教程,每一步操作都比较详细。 http://www.baderlab.org/Software/EnrichmentMap/Tutorial
-
GSEA软件和数据集下载 http://software.broadinstitute.org/gsea/downloads.jsp
-
原文对GSEA原理的讲解是很清晰的,可以读下,关键的内容也都摘录在第一个链接里。 https://www.ncbi.nlm.nih.gov/pubmed/16199517
以上是关于基因集富集分析 --- GSEA的主要内容,如果未能解决你的问题,请参考以下文章