R语言基因组数据分析可能会用到的data.table函数整理
Posted ywliao
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言基因组数据分析可能会用到的data.table函数整理相关的知识,希望对你有一定的参考价值。
版权声明:本文为博主原创文章,转载请注明出处
R语言data.table包是自带包data.frame的升级版,用于数据框格式数据的处理,最大的特点快。包括两个方面,一方面是写的快,代码简洁,只要一行命令就可以完成诸多任务,另一方面是处理快,内部处理的步骤进行了程序上的优化,使用多线程,甚至很多函数是使用C写的,大大加快数据运行速度。因此,在对大数据处理上,使用data.table无疑具有极高的效率。这里主要介绍在基因组数据分析中可能会用到的函数。
fread
做基因组数据分析时,常常需要读入处理大文件,这个时候我们就可以舍弃read.table,read.csv等,使用读入速度快的fread函数
fread(input, sep="auto", sep2="auto", nrows=-1L, header="auto", na.strings="NA", file,
stringsAsFactors=FALSE, verbose=getOption("datatable.verbose"), autostart=1L,
skip=0L, select=NULL, drop=NULL, colClasses=NULL,
integer64=getOption("datatable.integer64"),# default: "integer64"
dec=if (sep!=".") "." else ",", col.names,
check.names=FALSE, encoding="unknown", quote="\\"",
strip.white=TRUE, fill=FALSE, blank.lines.skip=FALSE, key=NULL,
showProgress=getOption("datatable.showProgress"), # default: TRUE
data.table=getOption("datatable.fread.datatable") # default: TRUE
)
input 输入的文件,或者字符串(至少有一个"\\n");
sep 列之间的分隔符;
sep2 分隔符内再分隔的分隔符,功能还没有应用;
nrow 读取的行数,默认-l全部,nrow=0仅仅返回列名;
header 第一行是否是列名;
na.strings 对NA的解释;
file 文件路径,再确保没有执行shell命令时很有用,也可以在input参数输入;
stringsASFactors 是否转化字符串为因子;
verbose 是否交互和报告运行时间;
autostart 机器可读这个区域任何行号,默认1L,如果这行是空,就读下一行;
skip 跳过读取的行数,为1则从第二行开始读,设置了这个选项,就会自动忽略autostart选项,也可以是一个字符,skip="string",那么会从包含该字符的行开始读;
select 需要保留的列名或者列号,不要其它的;
drop 需要取掉的列名或者列号,要其它的;
colClasses 类字符矢量,用于罕见的覆盖而不是常规使用,只会使一列变为更高的类型,不能降低类型;
integer64 读如64位的整型数;
dec 小数分隔符,默认"."不然就是","
col.names 给列名,默认试用header或者探测到的,不然就是V+列号;
encoding 默认"unknown",其它可能"UTF-8"或者"Latin-1",不是用来重新编码的,而是允许处理的字符串在本机编码;
quote 默认""",如果以双引开头,fread强有力的处理里面的引号,如果失败了就会用其它尝试,如果设置quote="",默认引号不可用
strip.white 默认TRUE,删除结尾空白符,如果FALSE,只取掉header的结尾空白符;
fill 默认FALSE,如果TRUE,不等长的区域可以自动填上,利于文件顺利读入;
blank.lines.skip 默认FALSE,如果TRUE,跳过空白行
key 设置key,用一个或多个列名,会传递给setkey
showProgress TRUE会显示脚本进程,R层次的C代码
data.table TRUE返回data.table,FALSE返回data.frame
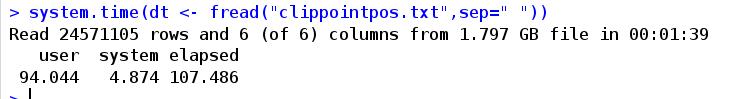
可见1.8GB的数据读入94秒,读入文件速度非常快
fwrite
对数据框数据进行处理后,需要保存到文件,我们就可以使用fwrite多线程写出,速度特别快
fwrite(x, file = "", append = FALSE, quote = "auto",
sep = ",", sep2 = c("","|",""),
eol = if (.Platform$OS.type=="windows") "\\r\\n" else "\\n",
na = "", dec = ".", row.names = FALSE, col.names = TRUE,
qmethod = c("double","escape"),
logicalAsInt = FALSE, dateTimeAs = c("ISO","squash","epoch","write.csv"),
buffMB = 8L, nThread = getDTthreads(),
showProgress = getOption("datatable.showProgress"),
verbose = getOption("datatable.verbose"))
x 具有相同长度的列表,比如data.frame和data.table等;
file 输出文件名,""意味着直接输出到操作台;
append 如果TRUE,在原文件的后面添加;
quote 如果"auto",因子和列名只有在他们需要的时候才会被加上双引号,例如该部分包括分隔符,或者以"\\n"结尾的一行,或者双引号它自己,如果FALSE,那么区域不会加上双引号,如果TRUE,就像写入CSV文件一样,除了数字,其它都加上双引号;
sep 列之间的分隔符;
sep2 对于是list的一列,写出去时list成员间以sep2分隔,它们是处于一列之内,然后内部再用字符分开;
eol 行分隔符,默认Windows是"\\r\\n",其它的是"\\n";
na,na 值的表示,默认"";
dec 小数点的表示,默认".";
row.names 是否写出行名,因为data.table没有行名,所以默认FALSE;
col.names 是否写出列名,默认TRUE,如果没有定义,并且append=TRUE和文件存在,那么就会默认使用FALSE;
qmethod 怎样处理双引号,"escape",类似于C风格,用反斜杠逃避双引,“double",默认,双引号成对;
logicalAsInt 逻辑值作为数字写出还是作为FALSE和TRUE写出;
dateTimeAS 决定 Date/IDate,ITime和POSIXct的写出,"ISO"默认,-2016-09-12,
18:12:16和2016-09-12T18:12:16.999999Z;"squash",-20160912,181216和20160912181216999;"epoch",-17056,65536和1473703936;"write.csv",就像write.csv一样写入时间,仅仅对POSIXct有影响,as.character将digits.secs转化字符并通过R内部UTC转回本地时间。前面三个选项都是用新的特定C代码写的,较快;
buffMB 每个核心给的缓冲大小,在1到1024之间,默认80MB;
nThread 用的核心数;
showProgress 在工作台显示进程,当用file==""时,自动忽略此参数;
verbose 是否交互和报告时间

dcast.data.table
和reshape2包的dcast一样, 这个函数用来重铸表格,并且再在大数据的处理上,比reshape2的内存更优化,函数效果如下

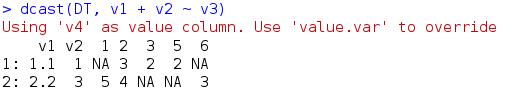
原表格 铸造后(v4作为value)
dcast(data, formula, fun.aggregate = NULL, sep = "_",
..., subset = NULL, margins=NULL,fill = NULL,
drop = TRUE, value.var = guess(data),
verbose = getOption("datatable.verbose"))
data 一个data.table;
formula 要铸造的表格的LHS~RHS格式;LHS和RHS可以是"..."和“.",其中"..."代表全部变量,"."代表无变量;
fun.aggregate 是否在铸造之前汇总,应提供函数list(比如mean,sum或者c(sum,mean)),默认length;
sep 铸造的时候连接字符变量的连接符,默认_;
subset 指定要铸造的子集;利用;
margins 函数尚不能应用(作者还没写好),预计设定编辑汇总方向;
fill 填充缺失值;
drop 设置成FALSE显示没有联合成功的行列
value.var 填充值的列,默认会猜测
现在我需要取数据DT的v1,v2两列相同的情况作为汇总的一类,对它们的v4值取平均,转换如下,


转换前 转换后
当然,上述过程也可以用data.table[ i , j , by ]语法做

但是如果我要将上述DT中的v3作为一个影响因素,作为tag,先按v1、v2汇总,再将对应的v4值分为v3=1和v3=2两类,查看v1、v2取值相同v3不同对应v4的情况,这个时候用dcast或者会更加方便,如下

melt
和reshape2包的melt一样,融合表格,这个是用C语言写的,处理速度更快。
处理前:

处理后:

melt(data, id.vars, measure.vars,
variable.name = "variable", value.name = "value",
..., na.rm = FALSE, variable.factor = TRUE,
value.factor = FALSE,
verbose = getOption("datatable.verbose"))
data data.table对象;
id.vars id变量组成的矢量,可以对应列号,也可以对应列名;缺失的话,非测量变量会被赋值;
measure.vars 测量变量组成的是矢量或者列表,可以对应列号和列名,也支持pattern函数,下面会提到,如果缺失,非id变量会被赋值;如果measure.vars和id.vars都没有赋予,全部非数字列会作为id.vars,剩余作为measure.vars;如果measure变量不是同一种类型,那么会被强制转换,等级如下list > character > numeric > integer > logical;
variable.name 测量变量列名,默认"variable";
value.name 融合后数据的数值列名;
na.rm 如果TRUE,移除NA值;
variable.factor 如果TRUE,变量列转化为因子;
verbose 如果TRUE,在工作台产生交互信息,默认options(datatable.verbose=TRUE)
对于前面的DT,我现在将f和d开头的列名的列作为测量变量,如下
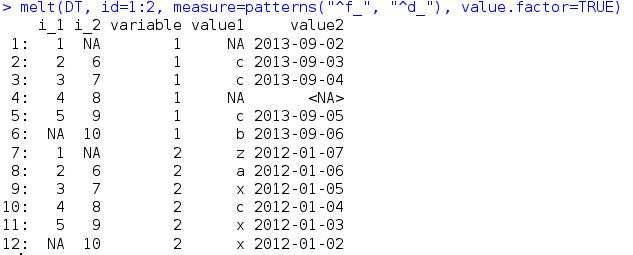
pattern函数下面会讲,这里再讲一下的是melt和dcast的联合使用,先用melt融合,再用dcast重铸
如下面例子
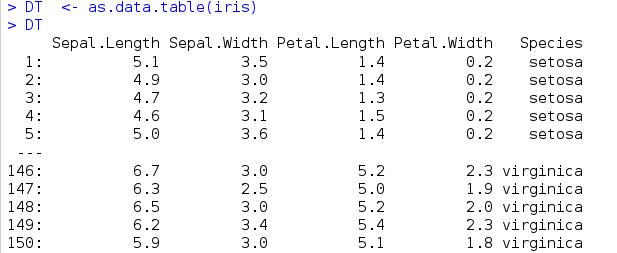
原DT
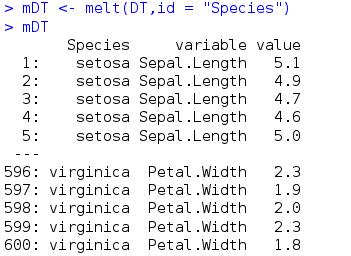

melt 后 再进行dcast后
其实上述过程用data.table [ i , j , by ]语法也可以
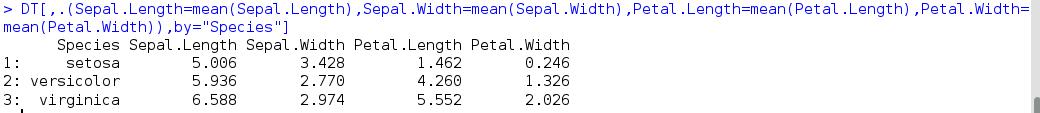
看个人需要吧,各种各样不同的方法都了解了以后,当你真正需要用到达到某个目的时,你的脑海里就会自动匹配上最佳的处理方法。
patterns
patterns是melt函数内部使用的函数,匹配正则表达式。melt的时候可以用正则去匹配列名
patterns(..., cols=character(0))
... 正则表达式集;
cols 要匹配的字符矢量;
例子在讲melt函数的时候已有
rbindlist
类似于data.frame的rbind,不过比rbind的速度更快,并且总是返回data.table。也有不同之处,一是use.names参数,可以指定是否使用相同列名bind,二是rbindlist可以使用在不知道对象名字的情况下,比如lapply(fileNames, fread) 。
rbindlist(l, use.names=fill, fill=FALSE, idcol=NULL)
l 对象列表,也可以分开写
use.names 如果TRUE, bind的时候匹配行名,默认FALSE,像rbind一样,直接bind,当时TRUE的时候,至少要有一个对象的一列要存在行名;
fill 如果TRUE,缺失的列用NA填充,这个时候bind的对象可以不同列数,并且use.names自动设为TRUE,这个时候至少要有一个对象的一列要存在行名;
idcol 产生一个index列,默认(NULL)不产生,如果idcol=TRUE,行名自动为.id,当然你也可以直接命名,比如idcol="id";



between
是data.table i 语法的扩展功能,between等同于x >= lower 并且 x <= upper 当incbounds设置为TRUE的时候,设置为FALSE的时候则是x > lower 并且 x < upper
between(x, lower, upper, incbounds=TRUE)
x %between% y
x 任意的可以排序的矢量,可以用"<="比较的
lower 较低的范围;
upper 较高的范围;
y 长度为2的矢量或者列表,y[1] 相当于lower,y[2] 相当于upper;
incbounds 如果TRUE意味着包括边界,即<=或者>= ,默认TRUE;
例如有基因组注释文件如下
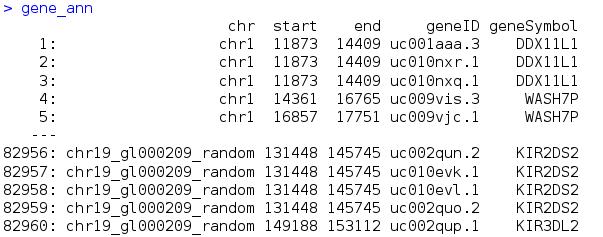
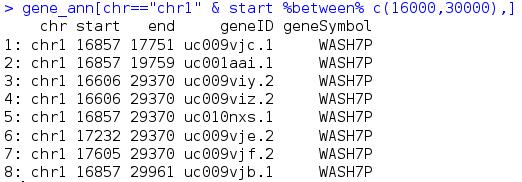
我想取出在chr1上,start在16000到30000之间的geneID,可以用beween
foverlaps
寻找重叠的区域,返回index对,x是数据很大但都是小区域的data.table,用来检索,y是检索用的资料,数据较小,都是大区域。



foverlaps(x, y, by.x = if (!is.null(key(x))) key(x) else key(y),
by.y = key(y), maxgap = 0L, minoverlap = 1L,
type = c("any", "within", "start", "end", "equal"),
mult = c("all", "first", "last"),
nomatch = getOption("datatable.nomatch"),
which = FALSE, verbose = getOption("datatable.verbose"))
x,y data.table,y需要设置key,x并不需要设置key;
by.x,by.y 用来计算重叠的列名或者列号的矢量,by.x和by.y的最后两列都应该对应各自的(x,y的)start和end区间列,并且start列应该总是小于end列,如果x设置了key ,by.x相当于key(x),否则by.x就默认key(y)。by.y默认key(y);
maxgap 设定两个区域空白区允许的最大值,参数尚不能使用;
minoverlap 设定两个区域最小的重叠区,参数尚不能使用;
type 设置重叠类型。默认any。可以设置为any,within,start,end和equal。equal尚不能使用。假设x,y区间分别为[ a,b ]和[ c,d ] , start 要求a==c , end要求b==d , within要求 a>=c 并且b <= d , equal要求a==c,b==d, 如果是any的话,只要c<=b 并且d>=a 就可以了;
mult 当y里面的多行都匹配x里面的行,mult=控制返回,默认all,也可以设置为"first”和last;
nomatch 默认nomatch=NA,无匹配返回NA,也可以设置为0,0不返回该行;
which 默认FALSE结果返回x和y行的联合,当是TRUE时,如果mult=“all”,返回两列,一列x列号,一列相对应的y,如果nomatch=NA,不匹配的返回y的NA,如果nomatch=0,则跳过该列,设置mult="first“,mult=”last"则最后返回x一样的行数;
verbose 当时TRUE的时候,工作台交互
chmatch
返回各字符串在第二个对象的首匹配位置,是match和%in%的加速版本。和fastmatch包的fmatch相比,各有优缺点。fmatch第一次匹配较慢,第二次匹配快,chmatch匹配虽然没有fmatch第二次匹配快,但是首次匹配也有较快的速度。
chmatch(x, table, nomatch=NA_integer_)
x %chin% table
x 字符矢量,需要去匹配的值;
table 字符矢量,匹配的目标;
nomatch 不匹配时返回的值,强制转化整型

好了,写到这里写的都有点累了,再介绍最后一个函数,有时候我们需要了解你写的这个脚本运行所花费的时间,这个时候保存开始运行时间和结束运行时间,再进行相减之类的好像有点麻烦,其实我们可以用这个timetaken函数
timetaken
timetaken(started.at)
started.at proc.time( )的结果

最后,写完这篇博客,timetaken断断续续大约一星期
参考文献
data.table manual: https://cran.r-project.org/web/packages/data.table/data.table.pdf
以上是关于R语言基因组数据分析可能会用到的data.table函数整理的主要内容,如果未能解决你的问题,请参考以下文章