R语言--聚类分析
Posted sakura-d
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言--聚类分析相关的知识,希望对你有一定的参考价值。
聚类算法:
- K-均值聚类(K-Means)
- K-中心点聚类(K-Meaoids)
- 密度聚类(Densit-based Spatial Clustering of Application with Noise,DBSCAN)
- 系谱聚类(Hierarchical Clustering)
- 期望最大化聚类(Expectation Maximization,EM)
- 其他算法
K-均值聚类算法
算法原理
K-均值聚类算法,是一种迭代算法,其采用距离作为判断对象之间相似性的指标,距离越近即相似度越高。这里的距离是欧式距离:m维空间中两点之间的真实距离。如二维空间中点(x1,y1)和点(x2,y2)的欧式距离为: \\(\\sqrt(x_2-x_1)^2+(y_2-y_1)^2\\) 。三维空间两点之间欧式距离为 \\(\\sqrt(x_2-x_1)^2+(y_2-y_1)^2+(z_2-z_1)^2\\) 。多维数据可以通过PCA(主成分分析)降维,或者直接计算欧式距离。
算法过程:
- 随机选取的K(簇个数,如游戏分为:顶级、高级、中级、低级,则k = 4)个样本作为起始中心点(簇)。
- 计算每个样本的点与各起始中心点的距离,将其余样本归入距离最近的簇,这就将所有样本完成了第一次分类。
- 确定当前类(簇)中样本坐标均值,作为新的起始中心点,然后计算各样本和各个新的起始中心点的距离,将样本按照距离归入距离最近的簇中。这是第二次分类。
- 确定新的起始中心点,把样本归类,依次循环迭代,直至所有样本归属类别不在变动(即最后确定了分类)。
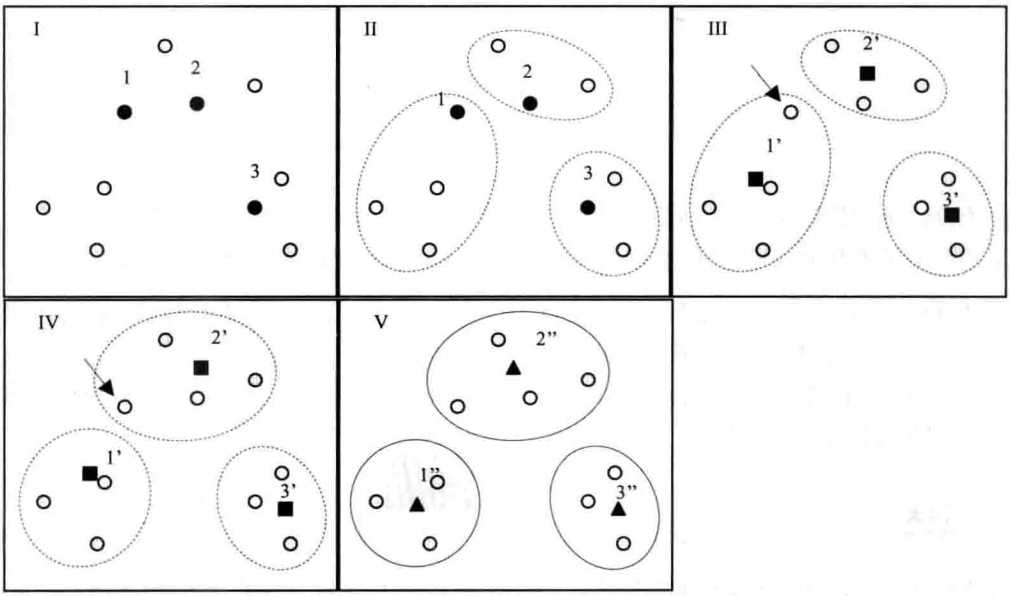
上图中10个样本,想分为3类,首先随机选取了3个样本点(黑色标记样本:1,2,3)(图Ⅰ)。然后计算其余样本点与这三个样本点的距离,然后根据距离分类(图Ⅱ)。计算当前分类中样本坐标均值,作为新的起始中心点(图Ⅲ,黑色小方块),然后计算样本与各新的起始中心点的距离进行分类(图Ⅳ)。然后图Ⅳ分类中再计算各类中样本坐标均值作为新的起始中心点(图Ⅳ中的黑色小方块),计算各样本和各新的起始中心点的距离,进行分类(图Ⅴ),发现图Ⅳ和图Ⅴ相同,即分类不在变化,标志分类完成。
R的实现
K-Means算法在R中的实现的核心函数为stats包中的kmeans()函数。
kmeans(x, centers, iter.max = 10, nstart = 1,
algorithm = c("Hartigan-Wong", "Lloyd", "Forgy",
"MacQueen"), trace=FALSE)
## S3 method for class 'kmeans'
fitted(object, method = c("centers", "classes"), ...)- x:待进行聚类分析的数据集
- centers:预设分类(簇)数目,即K值
- iter.max:最大迭代次数,默认值为10
- nstart:选择随机起始中心点的次数,默认值为1
- algorithm:该参数提供了四种参数选择,默认为Hartigan-Wong
- trace:该参数仅用于默认算法(Hartigan-Wong),如果设定为正(或TRUE),则生成算法进程相关的进程信息,如果数据量大的可能生成会生成跟多的进程信息。
实例
以iris数据集进行聚类分析,iris数据集:安德森鸢尾花卉数据集,包含150个样本(行),每个样本的5中特征向量(花萼长度,花萼宽度,花瓣程度,花瓣宽度,品种)。
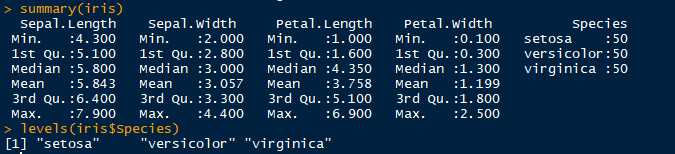
观察数据发现,有三个品种,每个品种为50个样本数据
kc <- kmeans(x = iris[1:4],centers = 3) # 以前四个特征数据进行聚类,分成3类。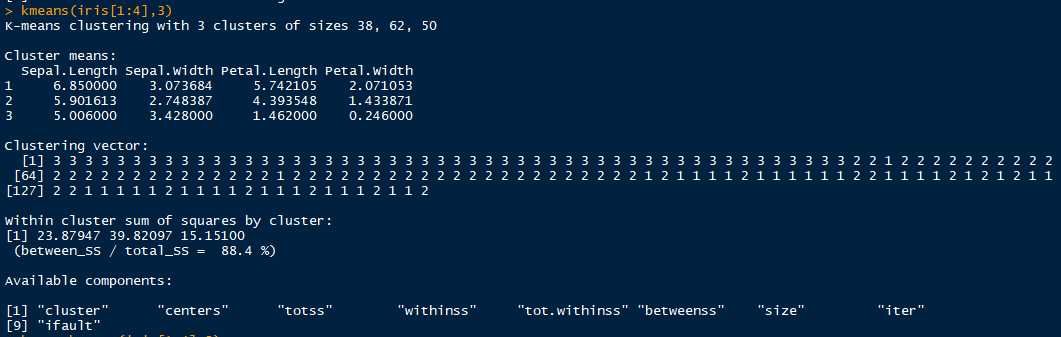
分成3类,第一类:38个样本;第二类:62个样本;第三类:50个样本。根据源数据可知,只有一类分类完全正确,有12个样本分类错误。
fitted(kc) # 查看最终分类后,各类的样本坐标均值,即该类的起始中心点
# 结果:
Sepal.Length Sepal.Width Petal.Length Petal.Width
1 5.006000 3.428000 1.462000 0.246000
3 5.901613 2.748387 4.393548 1.433871
2 6.850000 3.073684 5.742105 2.071053table(iris$Species,kc$cluster) # table函数:通过交叉分类构建因子水平频数列联表。
# 结果:
1 2 3
setosa 50 0 0
versicolor 0 2 48
virginica 0 36 14列联表可以比较直观的看出分类的具体情况,从上面可以看出:setosa50个样本完全分类正确;把14个virginica样本分为versicolor,同时把2个versicolor分为virginica
可视化
plot(iris[c("Sepal.Length","Sepal.Width")],col = kc$cluster,pch = as.integer(iris$Species)) 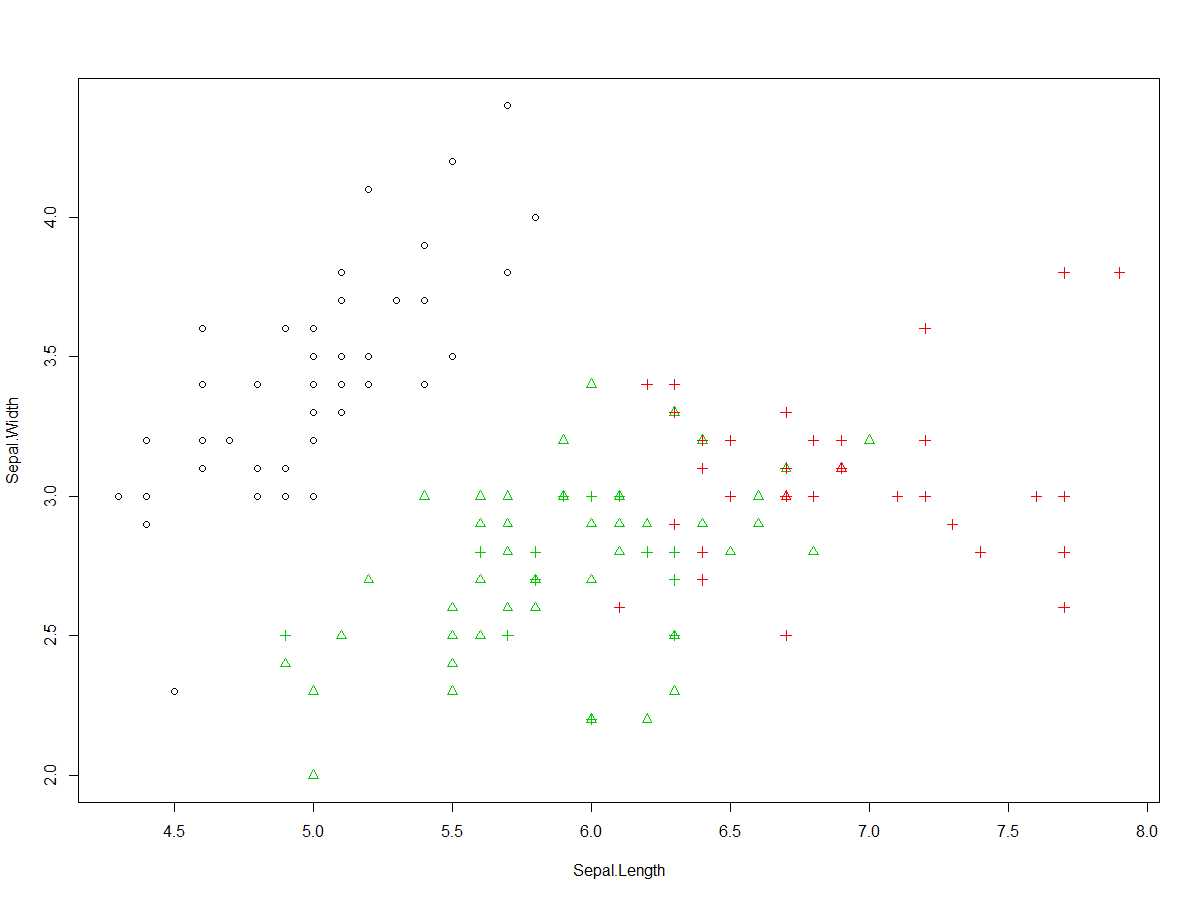
上面可视化:以花萼长度和花萼宽度散点图,以形状对源数据分类进行分组显示,以颜色作为聚类分组显示,即原来每种50个样本,如果分类正确则每种颜色,每种形状点有50个。
圆点:setosa;三角形:versicolor;十字形:virginica
白色:setosa;绿色:versicolor;红色:virginica
从图中可以看到:
圆点、白色一致,即setosa分类完全正确
绿色三角形48个,红色三角形2个,即versicolor有48个分类正确,有两个被归入virginica
红色十字形36个,绿色十字形14个,即virginica种的36个样本正确归类为virginica,有14样本被归类为versicolor
以上是关于R语言--聚类分析的主要内容,如果未能解决你的问题,请参考以下文章
R语言聚类分析之基于划分的聚类KMeans实战:基于菌株数据
R语言聚类分析之基于划分的聚类KMeans实战:基于葡萄酒数据