易基因:禾本科植物群落的病毒组丰度/组成与人为管理/植物多样性变化的相关性 | 宏病毒组
Posted E-GENE
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了易基因:禾本科植物群落的病毒组丰度/组成与人为管理/植物多样性变化的相关性 | 宏病毒组相关的知识,希望对你有一定的参考价值。
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
现代农业通过简化生态系统、引入新宿主物种和减少作物遗传多样性来影响植物病毒的出现。因此,更好理解农业生态中种植和未种植群落中的病毒分布,以及它们之间的病毒交换至关重要。2023年03月14日,《Microbiol Spectr》杂志发表了题为“Long-Term Anthropogenic Management and Associated Loss of Plant Diversity Deeply Impact Virome Richness and Composition of Poaceae Communities”的研究论文,研究通过对三种禾本科(Poaceae)群落类型进行宏病毒组测序分析,揭示了长期人为管理和植物多样性破坏会严重影响禾本科植物群落的病毒组丰度和组成。

标题:Long-Term Anthropogenic Management and Associated Loss of Plant Diversity Deeply Impact Virome Richness and Composition of Poaceae Communities(长期人为管理和植物多样性破坏会严重影响禾本科植物群落的病毒组丰度和组成)
期刊:Microbiology Spectrum
影响因子:IF 9.043
发表时间:2023.03.14
研究摘要:
本研究通过表征三种不同物种丰富度和管理强度梯度的禾本科群落类型的病毒组(从管理强度高的作物单一种植到管理强度低的物种丰富度高),从而推进对这一领域的基本理解。研究人员在两年多的时间里对950株草地野生和种植的禾本科植物进行大规模研究,将非靶向病毒组(virome)分析与病毒种类水平结合,并靶向检测三种植物病毒。
病毒宏基因组学的深度测序揭示了:
(i)多样化且基本未知的禾本科植物病毒组(至少51种病毒物种或分类群),具有持久性病毒富集;
(ii)群落内的病毒组丰度随着物种丰富度的增加而增加;
(iii)病毒组丰度在不同时间比较稳定,但病毒种类变异性大;
(iv)植物群落和物种之间病毒流行、多种感染和空间分布的对比模式。
研究结果突出了自然界中植物病毒群落的复杂结构,并表明了人为管理对病毒分布和流行的影响。
研究重要性:
由于先前研究主要在种植植物中研究病毒,因此对人为管理较少植被中的病毒多样性和生态学、人为管理和农业对病毒组组成的影响知之甚少。由于禾本科(grass family)植物分布在世界各地的农业生态梯度中,对粮食安全和保护至关重要,并且可能被多种病毒感染,因此以禾本科植物为主的群落为研究这些生态问题提供了宝贵的机会。本研究利用多层次分析,同时考虑植物群落、单株植物、病毒种类和单倍型遗传多样性,以扩大对禾本科植物病毒组的理解,并评估了研究区域农业生态中宿主-寄生生物丰富度的关系。研究揭示了草地多样性和土地利用对病毒群落组成及其生活史策略的影响,表明了管理较少的草地群落中植物-病毒互作的复杂性,如与理论预测相比,病毒流行率、多重病毒合并感染的比例更高。
研究方法
在2017和2018年两年的时间,对相距10公里的3个地点(Antheit、Héron、Latinne)分别抽样采集麦田(植物物种丰富度低,管理强度高)、集约化牧场(物种丰富度和管理强度均中等)和自然草地(物种丰富度高,管理强度低)的禾本科植物各50株;并在2018年采集了附近一块具有真菌感染症状的麦田中的50株禾本科植物。对一共19次采集的样品分别进行病毒样颗粒富集后提取总核酸(包括DNA和RNA病毒),进行宏病毒组测序分析。
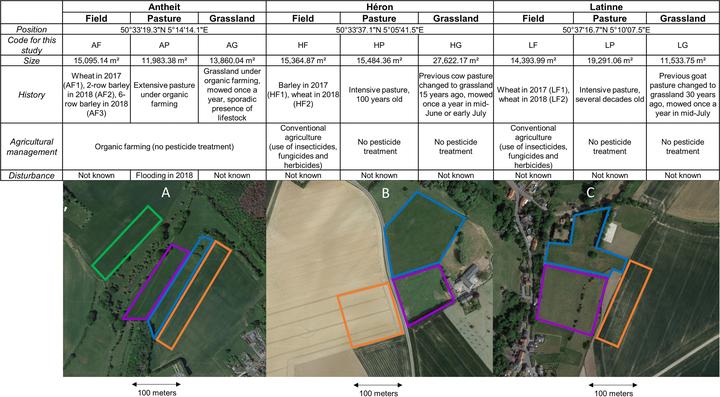
图1:所研究的不同群落和地点的信息表(GPS位置、代码、规模、历史、农业管理和干扰因素)。下图为Antheit(a),Héron(B)和Latine(C)位置,主要分析了三个相邻的生态系统:大麦或麦田(橙色)、集约化牧场(紫色)和物种丰富度高的草地(蓝色)。2018年在Antheit对另一块 6行的大麦田进行采样(绿色)。
结果
(1)病毒宏基因组测序揭示了禾本科植物中多样化且大部分未知的病毒组
为评估每个群落中的病毒组,采样了24个禾本科物种的950株植物,并对每个群落的50个样本使用病毒宏基因组测序方法(virion-associated nucleic acids ,VANA)。文库平均包含17.9%的病毒reads(从0%~78%)。由于在禾本科植物样本中检测到的外来靶序列(PLRV、BSV)平均占文库中总reads的0.01%,因此观察到低水平的交叉样本污染,以此作为本研究的检测阈值(不检测低于此水平病毒)。组装了51个共有植物病毒基因组(1496 - 13876 核苷酸(nt)长度),覆盖了在相应属或科中描述ORF中的47个。研究鉴定出四个不完整的病毒基因组(1497nt - 6785nt长度):Closterovirus病毒、Rymovirus病毒、Varicosavirus病毒、Amalgavirus病毒。这51种RNA病毒被比对到代表21个病毒属的16个病毒家族(表1)。分析结果表明,超2/3的禾本科植物病毒组在很大程度上仍然未知(n=37),这些新病毒物种主要是在所有三种群落类型中均发现的持久性病毒和真菌病毒(3种novel alphachrysoviruses、10种novel Partitiviruses和13种novel totiviruses),不同时间的病毒组比较显示,77%的病毒种类在两个采样年份都被检测到(植物病毒为74%,真菌病毒为79%)。当只考虑长期植物群落(牧场和草地)时,这一值增加到85%。
表1:在禾本科植物群落(麦田、放牧牧场和割草草地)中检测到的不同病毒科、属和种
(2)禾本科植物群落与病毒组丰度或组成之间的关系
HTS的初步结果揭示了植物群落中病毒组丰度的对比水平。麦田、牧场和草地的病毒种类群丰度存在显著差异(在病毒科、属和种水平上,P<0.001,Kruskall-Wallis检验)。在麦田中发现的病毒种类很少或根本没有,而在更多样化的群落中观察到病毒组多样化,在草地中检测到的病毒种类多达22种(图2)。牧场和草地在科和属水平上的病毒组丰度没有显著差异(P=0.456和0.419,Mann-Whitney检验)。但在物种水平上,草地的多样性略高于牧场(Mann-Whitney检验,P=0.078,排除HG1时P=0.010)。使用线性回归模型来显示植物物种丰富度和病毒组丰度之间的关系(病毒组=0.60 + 0.62植物种类,R2=74.3%,P=0.000),病毒属丰富度(病毒属=0.55+0.82植物种类,R2=72.9%,P=0.000),以及病毒物种丰富度(病毒物种=0.22+1.57植物种类,R2=76.0%,P=0.000)(图2)。
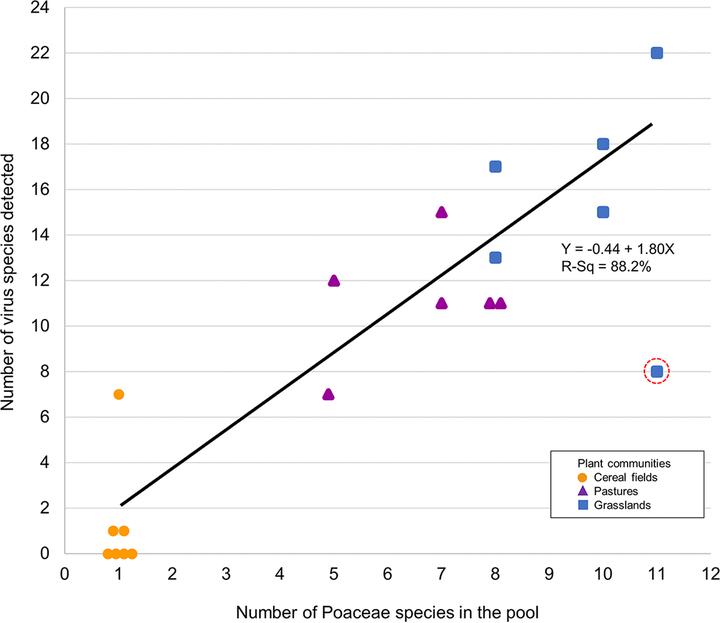
图2:2年间研究中植物群落(橙色圆麦田、紫色三角形牧场、蓝色方块草地)的禾本科植物物种丰富度与病毒种类丰富度之间的关系。2017年研究的Héron草地采用不同的割草管理,以红色圆圈标记。排除Héron草地进一步将R2值从76.0%提高到88.2%。
禾本科植物群落中的病毒组组成通过对每个草地群落中鉴定的病毒种类进行网络分析来可视化(图3),并对草地和牧场的病毒和植物两个维度进行聚类分析,以突出任何群落聚类或病毒共生模式(图4)。
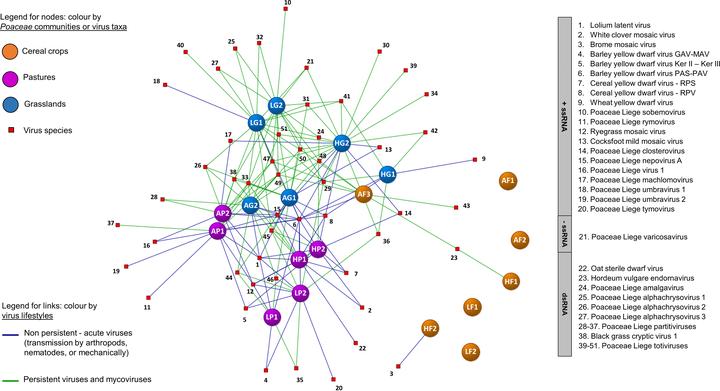
图3:对三个地点2年研究的禾本科植物群落(麦田、牧场、草地)中鉴定的病毒物种进行网络分析。病毒种类(红色方块)、植物群落(彩色球)。根据每个病毒物种(非持久性病毒、持久性病毒和真菌病毒)的假定方式着色。
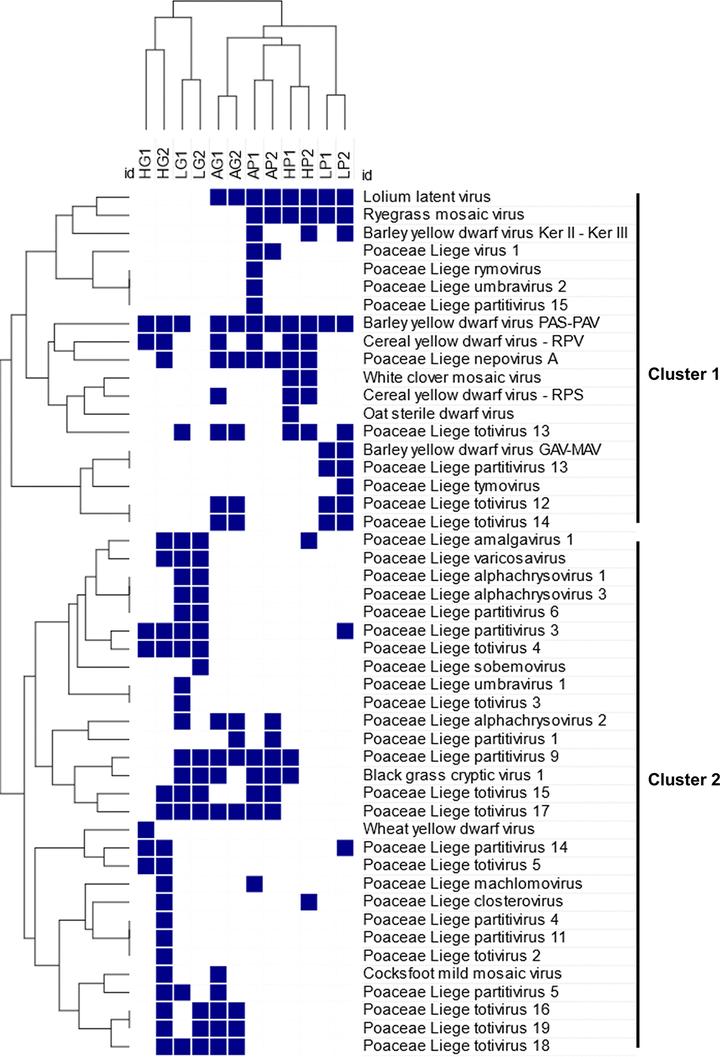
图4:植物和病毒两个维度的分层聚类分析。列表示2017年(1)和2018年(2) 两年间的不同地点(Antheit(A),Héron(H)和Latine(L))两个不同的非种植禾本科植物群落分析(牧场(P)和草地(G))。行表示检测到的不同病毒种类。右侧树状图突出显示了两组主要的共生病毒组。
(3)在人为管理较少的植物群落中,病毒流行率和多重感染率更高
为完成禾本科植物库病毒组组成提供的第一个生态信息,研究分析了3个样品地块内的病毒流行率、病毒共感染和病毒分布。对2018年在Antheit地点采集的单株植物HTS检测到的三种病毒进行病毒检测:barley yellow dwarf virus-PAV病毒 (BYDV-PAV,所有群落中均检测到)和两种新secovirids病毒(Poacee Liege nepovirus A病毒(PoLNVA)和Poacee Liege virus 1病毒(PoLV1),在牧场和草地中检测到)。分析了草地、牧场、6行大麦田和Lolium perenne、Poa trivilis两个优势物种的单株植物。分析结果表明,BYDV-PAV和PoLV1在植物群落类型和物种中具有不同的流行模式(图5A)。在麦田中没有出现高的病毒流行率,BYDV-PAV病毒流行率仅为6%。但在非种植区域的病毒流行率较高,特别是BYDV-PAV病毒和PoLNVA病毒的共感染(高达50%的植物在草地中共感染)。不过,三种病毒的多重感染相对较少(0%-7%)(图5B)。
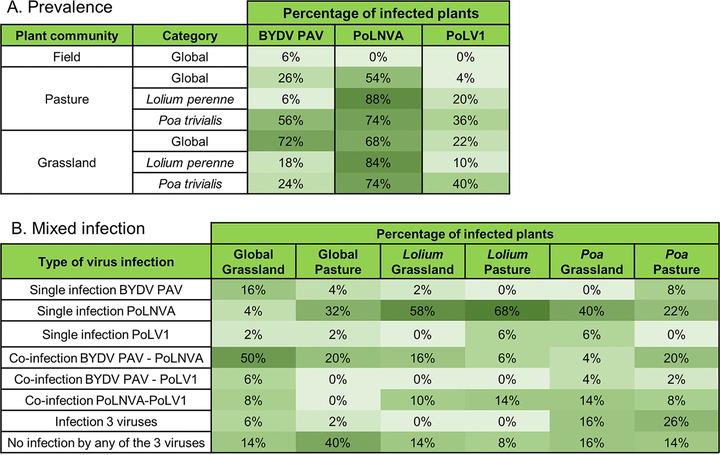
图5:(A)2018年在Anthit地点的植物群落和物种中观察到的BYDV-PAV和两种新物种(PoLNVA和PoLV1)的病毒流行率。
(B) 2018年在Antheit地点的非种植植物群落(草地和牧场,称为Global)和两种非种植禾本科植物(多年生Lolium perenne和Poa tivialis)中BYDV-PAV、PoLNVA和PoLV1的单一和多重感染百分比。
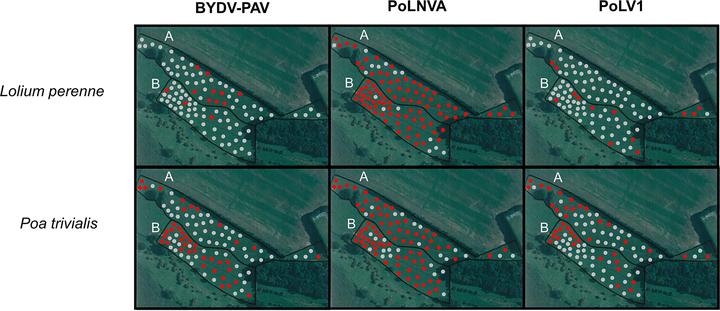
图6:在Antheit草地(A)和牧场(B)中多年生Lolium perenne和Poa trivilis的受感染植物(BYDV-PAV、PoLNVA和PoLV1)的地理分布。灰点和红点分别表示未感染和感染的植物。
(4)自然草地中的大型病毒遗传结构
同一植物群落内病毒序列的年际比较分析表明,许多病毒物种具有高基因组水平的遗传稳定性。例如在两个采样年份之间的Anthit牧场中,观察到黑麦草花叶病毒(ryegrass mosaic virus,RMV)和PLNA的核苷酸同源性分别为99.4%和99.8%。然而,在Lolium潜伏病毒(Lolium latent virus,LLV;Lolavirus属,Alphaflexiviridae科)发现更多变异性。对本研究中检测到的LLV分离株的RdRp区域的共有成对同源性分析进一步证实了其遗传多样性。对LLV-RdRp序列进行系统发育分析(图7),结果揭示了LLV的三个主要簇。在所研究2年间的同一地块中(如AP1-C1和AP2-C5、HG1-C1和HG2-C1)、在相同地点的牧场和草地(如AG1-C3和AP1-C4、HP2-C2和HG2-C2)、在不同位点(如AG1-C2、HP2-C1和LP2-C2)、给定的图和/或位点(如HP1-C1、LP1-C1)仅检测一次,均检测到密切相关的RdRp序列。此外,在同一位点中同时观察到非常不同的RdRp序列(如AG1-C1、AG1-C2和AG1-C3),其中一些与在其他位点中鉴定的序列密切相关(如AG1-C2、HP2-C1和LP2-C2)。
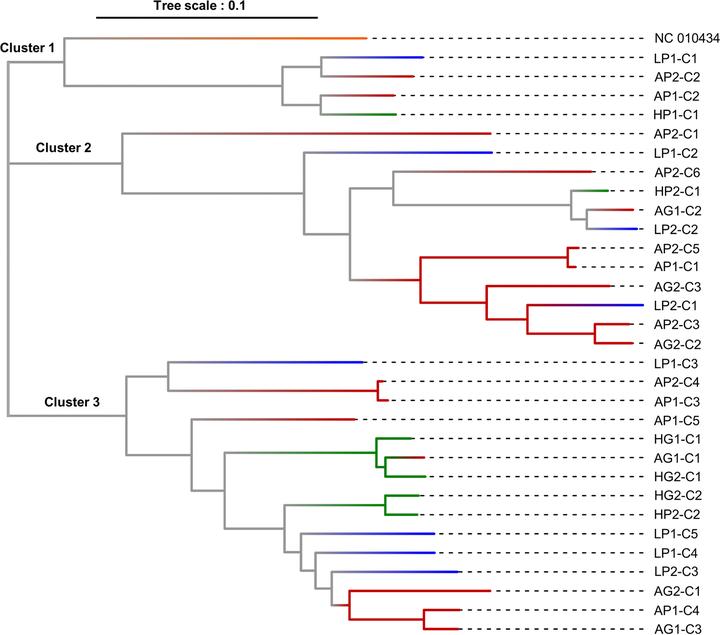
图7: 在2017(1)和2018(2)两年间,不同地点(红色Anthit[A]、绿色Héron[H]和蓝色的Latine[L])和群落(草地[G]和牧场[P])的L. perenne中的LLV病毒RdRp区域共有序列(contigs de novo assembled (Cx))系统发育树(最大似然法,Tamura-Nei模型,1000个bootstraps,70%阈值)。样品与LLV的NCBI对照比较(NC_010434)进行比较,显示为橙色。
易基因小结:
本研究中使用的宏病毒组测序方法可以扩展到其他地区、不同环境梯度和气候的更多禾本科植物物种以及其他植物科(例如茄科、豆科)。可以更广泛地概述混合物种群落中存在的病毒群落,并有助于理解不同农业生态的植物病毒生态学,该领域仍处于起步阶段。
关于易基因宏病毒组测序技术介绍
病毒宏基因组学(Meta-virome)是在宏基因组学理论的基础上,结合现有的病毒分子生物学检测技术而兴起的一个新的学科分支,是某类样本中所有病毒(virus)或病毒类似物(virus-like-particle)及其所携带遗传信息的总称。宏病毒组直接以环境中所有病毒的遗传物质为研究对象,能够快速准确的鉴定出环境中所有的病毒组成,在病毒发现、病毒溯源、微生物预警等研究方面具有重要作用。宏病毒研究可应用于人或动物肠道或者血液样本、海洋、土壤等的研究,用以挖掘潜在的对人类和环境的危害。
易基因科技同丹麦哥本哈根大学知名微生物课题组联合开发高效的宏病毒组颗粒富集方法,可对微量样本进行病毒颗粒富集,并保证病毒活性及高均质性,为宏病毒组学、噬菌体与细菌之间的互作关系等研究提供优良技术支持,保驾护航!
技术优势:
- 对样本进行病毒颗粒富集,实现微量样本中的病毒组进行有效分离。
- 对噬菌体不造成损伤,最大程度保证了病毒活性,适用于下游FVT研究。
- 相较于传统的PEG,CsCl等方法,均质性高,噬菌体偏好性低。
- 全面的数据分析策略,定制的多组学关联数据挖掘。
研究方向:
宏病毒组学研究在病毒发现、病毒溯源、微生物预警等研究方面具有重要作用:
- 农业领域:环境污染、生态系统破坏和作物残留化学物质
- 生态环境领域:调控寄主群落结构、参与元素地球化学循环、基因水平移动媒介
- 医药领域:噬菌体和噬菌体疗法的研究、人体肠道病毒组研究
易基因提供全面的微生物组学研究解决方案,详询易基因:0755-28317900
参考文献:
Maclot F, Debue V, Malmstrom CM, Filloux D, Roumagnac P, Eck M, Tamisier L, Blouin AG, Candresse T, Massart S. Long-Term Anthropogenic Management and Associated Loss of Plant Diversity Deeply Impact Virome Richness and Composition of Poaceae Communities. Microbiol Spectr. 2023 Mar 14:e0485022.
相关阅读:
代谢组与微生物联合分析实战
微生物组测序 (主要指扩增子测序、全长扩增子测序与宏基因组测序)可提供细菌构成、基因丰度和功能性信息,可以解决“who is there”(那儿有谁)和“what are they doing”(在干嘛)的问题。而代谢组学是研究生物体中代谢产物变化的科学,可以解决“what have really happened”(究竟发生了什么)的问题。生物科学研究过程复杂,单独和片面的单一组学无法解释清楚生物学问题,多组学就显得尤为重要。近年来,随着微生物组学研究的不断发展和持续火热,越来越多的研究者开始将微生物组学和代谢组学联合起来,从物种、基因以及代谢产物等水平共同解释科学问题,更好地理解疾病病变过程及机体内物质的代谢途径。还有助于发现疾病的生物标记以更进一步应用于临床辅助诊断。代谢和微生物的联合分析可以有多种方法,今天主要介绍相关性分析和协惯量分析。
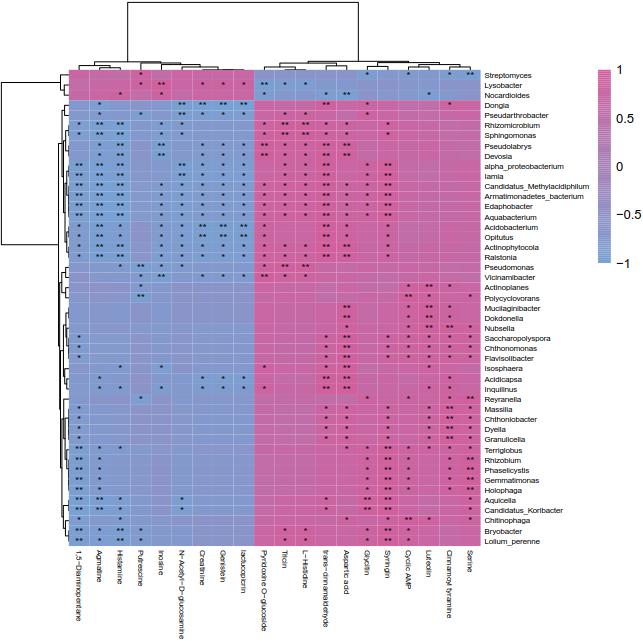
文章《Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention》中揭示了中国肥胖人群的肠道菌群组成,并指出了肥胖及减肥干预后肠道菌群和血清代谢物改变。其中MLG(metagenomic linkage groups,宏基因组连锁群)和代谢物联合分析就运用了相关性分析和协惯量分析(如下图)。
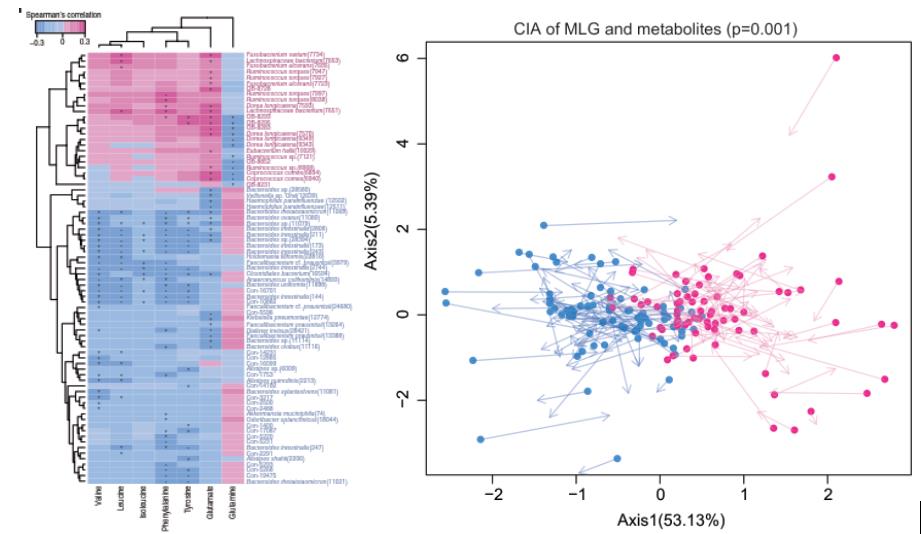
这里使用R基于示例数据绘制类似的图,需要提前安装psych、ade4、pheatmap等R包。另外使用TakeColor软件可直接获取文章图中的配色。使用的示例数据为6个样本的数据,对照处理各3个样本。

#代谢组数据,行为代谢物共20个,列为样本名
>dim(metabdata)
[1] 20 6
#微生物数据,行为微生物共47个,列为样本名
>dim(microdata)
[1] 47 6
#加载相关性计算的包
>library(psych)
#使用spearman计算相关性和p值
>cor_res<-corr.test(cbind(t(metabdata),t(microdata)),method='spearman',adjust='none',ci=F)
#显示数据
> dim(cor_res$p)
[1] 67 67
> tail(cor_res$r[,1:3])
Cyclic AMP Tricin Genistein
Pseudomonas 0.7714286 0.9428571 -0.7714286
Ralstonia 0.6667367 0.8406680 -0.8986451
Rhizobium 0.8285714 0.6571429 -0.7142857
Saccharopolyspora 0.8406680 0.6667367 -0.7247138
Terriglobus 0.8986451 0.7247138 -0.6667367
Vicinamibacter 0.7142857 0.8857143 -0.8285714
#提取相关性和p值
> corCmat<-cor_res$r[(nrow(metabdata)+1):nrow(cor_res$r),1:nrow(metabdata)]
> corPmat<-cor_res$p[(nrow(metabdata)+1):nrow(cor_res$p),1:nrow(metabdata)]
#绘制热图
> library(pheatmap)
> library(RColorBrewer)
#标记显著当p<0.01 显示2个星号‘**’,0.01<p<0.05 时显示一个星号‘*’
>annolabel<-matrix('',nrow(corPmat),ncol(corPmat))
> annolabel[corPmat<0.01]<-'**'
> annolabel[corPmat<0.05 & corPmat>=0.01]<-'*'
> colnames(annolabel)<-colnames(corPmat)
> rownames(annolabel)<-rownames(corPmat)
pheatmap(mat=corCmat,
display_numbers=annolabel,
number_color='black',
cluster_cols=T,
cluster_rows=T,
color=colorRampPalette(c('#7AA1D3','#D2609E'))(100),
border_color=F,
show_rownames=T,
show_colnames=T,
fontsize_number=8,
fontsize_row=6,
fontsize_col=6,
scale='none')
(向左查看更多内容)
协惯量分析:
#加载包
> library(ade4)
#进行协惯量分析
> microdata_dudi <- dudi.pca(t(microdata), scale = TRUE, scan = FALSE, nf = 2)
> metabdata_dudi <- dudi.pca(t(metabdata), scale = TRUE, scan = FALSE, nf = 2)
> coin<- coinertia(metabdata_dudi,microdata_dudi, scan = FALSE, nf = 2)
#基于特征值计算贡献
>Axis1<-coin$eig[1]/sum(coin$eig)*100
>Axis2<-coin$eig[2]/sum(coin$eig)*100
#绘制样本空间图,展示样本之间的差异
>metabS<-coin$mX
>microS<-coin$mY
>plot(microS[,1],microS[,2], xlim = c(-1.5,1.5), ylim = c (-2,2),
pch=20,col=c(rep('#4890CD',3),rep('#EC2B91',3)),
xlab=sprintf('Axis1(%.2f%%)',Axis1),ylab=sprintf('Axis2(%.2f%%)',Axis2))
>arrows(x0=microS[,1],y0=microS[,2],
x1=metabS[,1],y1=metabS[,2],
col=c(rep('#4890CD',3),rep('#EC2B91',3)),length=0.1)
(向左查看更多内容)
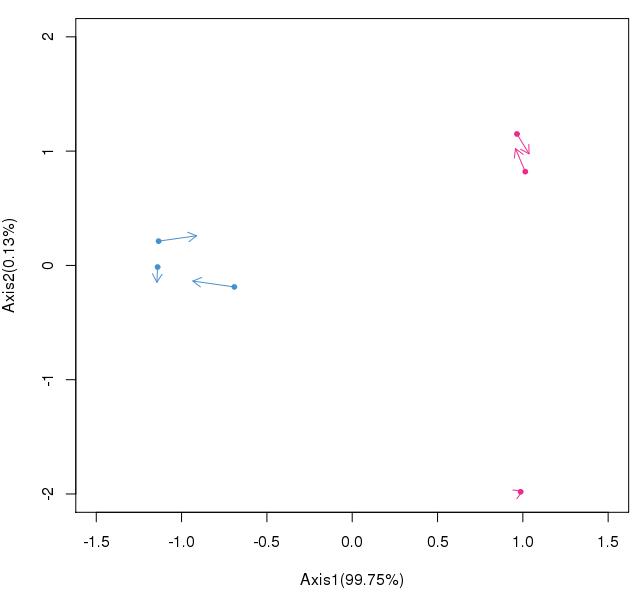
图中的蓝色和红色分别代表不同的样本分组即对照和处理,基于此分析可直观展示出样本间的差异。
对于没有R基础的同学可能会觉得困难,但是没关系,目前百迈客云已有代谢组与微生物联合分析以及个性化相关的绘图,只需要点点鼠标便可得到结果。
文:QG
排版:市场部
参考文献:
Liu R, Hong J, Xu X, et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention[J]. Nature medicine, 2017, 23(7): 859.
以上是关于易基因:禾本科植物群落的病毒组丰度/组成与人为管理/植物多样性变化的相关性 | 宏病毒组的主要内容,如果未能解决你的问题,请参考以下文章
易基因:DNA甲基化和转录组分析揭示野生草莓干旱胁迫分子调控机制|植物抗逆
易基因:2023年植物表观转录组研究的最新进展(m6A+m5C)|深度综述
易基因:ChIP-seq等揭示热休克转录因子A1b调控植物高温胁迫响应的分子机制|应激反应