使用Sentieon加速甲基化(WGBS)分析
Posted insvast
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了使用Sentieon加速甲基化(WGBS)分析相关的知识,希望对你有一定的参考价值。
全基因组甲基化测序(WGBS)是一种研究DNA甲基化的方法,以全面了解在基因组水平上的表观遗传变化。在进行WGBS数据分析时,通常需要使用专门的比对工具,因为这些工具需要能够处理亚硫酸盐转化后的数据。
以下是四个不同的WGBS比对分析流程:
- Bismark:Bismark是一个基于Bowtie2或HISAT2比对器的流行WGBS分析工具。它允许处理双链亚硫酸盐转化测序数据,并提供甲基化位点的检测和分析。
- BitmapperBS:BitmapperBS是一个专门为亚硫酸盐转化测序数据设计的高效比对器。它可以处理双链测序数据,并提供甲基化位点的检测和分析功能。
- BSseeker2:BSseeker2是一个用于WGBS数据分析的比对工具。它可以处理单链和双链亚硫酸盐转化测序数据,并支持Bowtie, Bowtie2和SOAPaligner作为比对器。BSseeker2提供了甲基化位点检测和甲基化水平计算等功能。
- BWA-Meth:BWA-Meth是一个基于BWA的比对工具,专门用于处理WGBS数据。它提供了处理双链亚硫酸盐转化测序数据的功能,并可以进行甲基化位点检测。
这四种分析流程各自具有不同的特点和优势,选择哪个流程取决于研究需求、计算资源以及期望的分析速度和准确性。实际应用中,可以尝试比较这些流程的结果,以找到最适合您需求的解决方案。
WGBS甲基化分析流程加速方案
Sentieon BWA + MethyDackel
在甲基化分析中,Sentieon软件可以与其他工具结合使用以提高分析速度和准确性。在这种情况下,Sentieon BWA被用来替换原始的BWA-mem,与MethyDackel结合,建立起Sentieon BWA-Meth流程。
在这个流程中,Sentieon BWA首先负责处理亚硫酸盐转化后的测序数据进行高效的序列比对。由于Sentieon BWA的优化,比对速度和准确性得到了提高,同时减少了计算资源的消耗。
接下来,MethyDackel被用于从Sentieon BWA的比对结果中提取甲基化信息。MethyDackel能够检测甲基化位点,计算甲基化水平,并生成甲基化状态的统计和可视化结果。
通过结合Sentieon BWA和MethyDackel,Sentieon BWA-Meth流程能够为全基因组甲基化分析提供一个高效且准确的解决方案。这使得研究人员可以更快地分析甲基化数据,更有效地挖掘潜在的生物学意义。
具体加速流程
Sentieon处理甲基化数据的过程可以概括如下:
-
Sentieon甲基化分析流程:
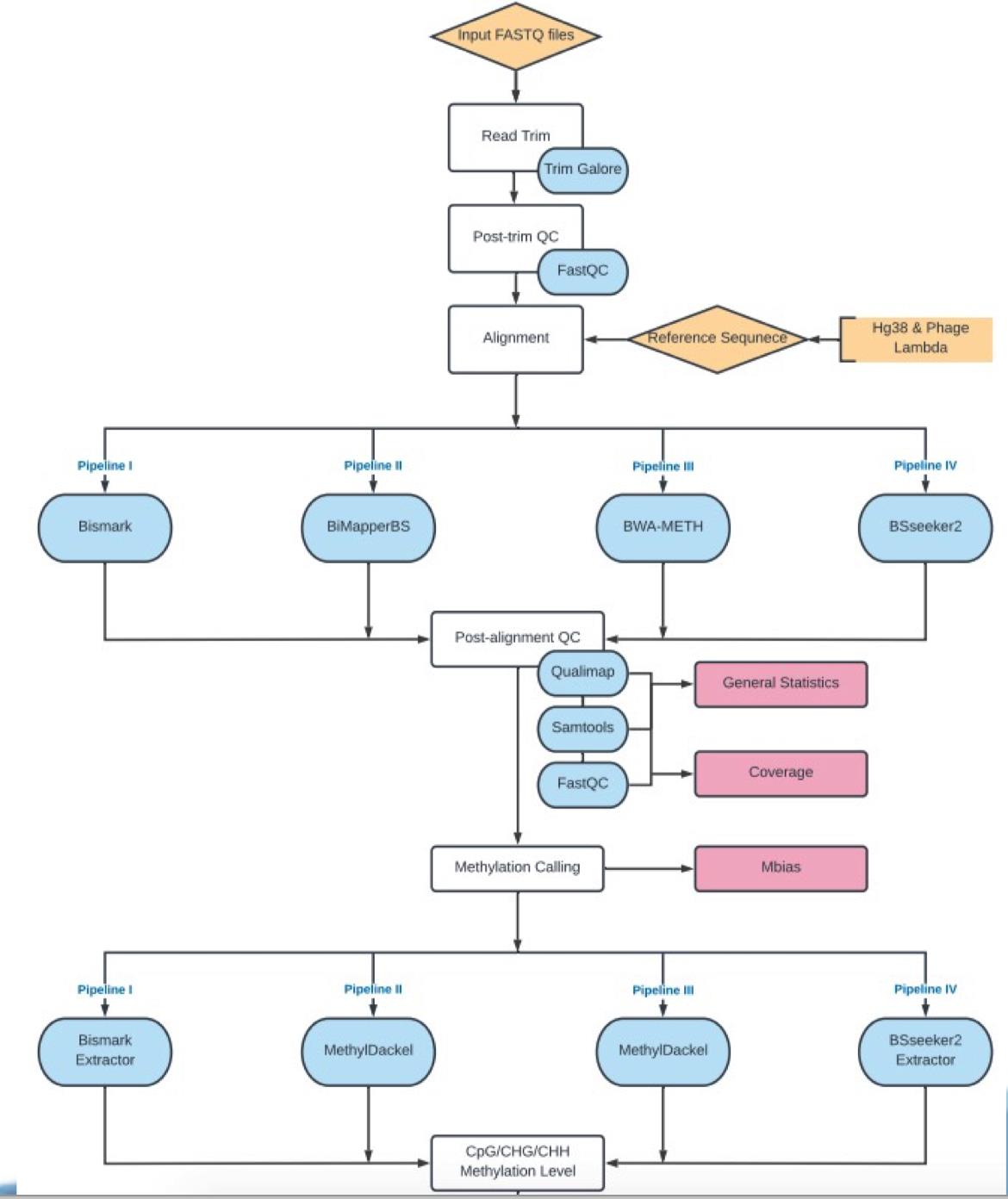
- 使用EpiQC研究中的全基因组甲基化测序数据(doi:https://doi.org/10.1101/2020.12.14.421529)。
- 数据预处理:读取修剪、质量控制。
- 使用四种不同的分析流程进行比对,包括Bismark、BitmapperBS、BSseeker2和BWAMeth。
- 使用Sentieon BWA替换原始的BWA-mem,并与MethyDackel结合,建立Sentieon BWAMeth流程。
- 比对后处理:使用不同的模块进行甲基化位点调用和CpG甲基化水平识别。
-
甲基化映射速度比较:
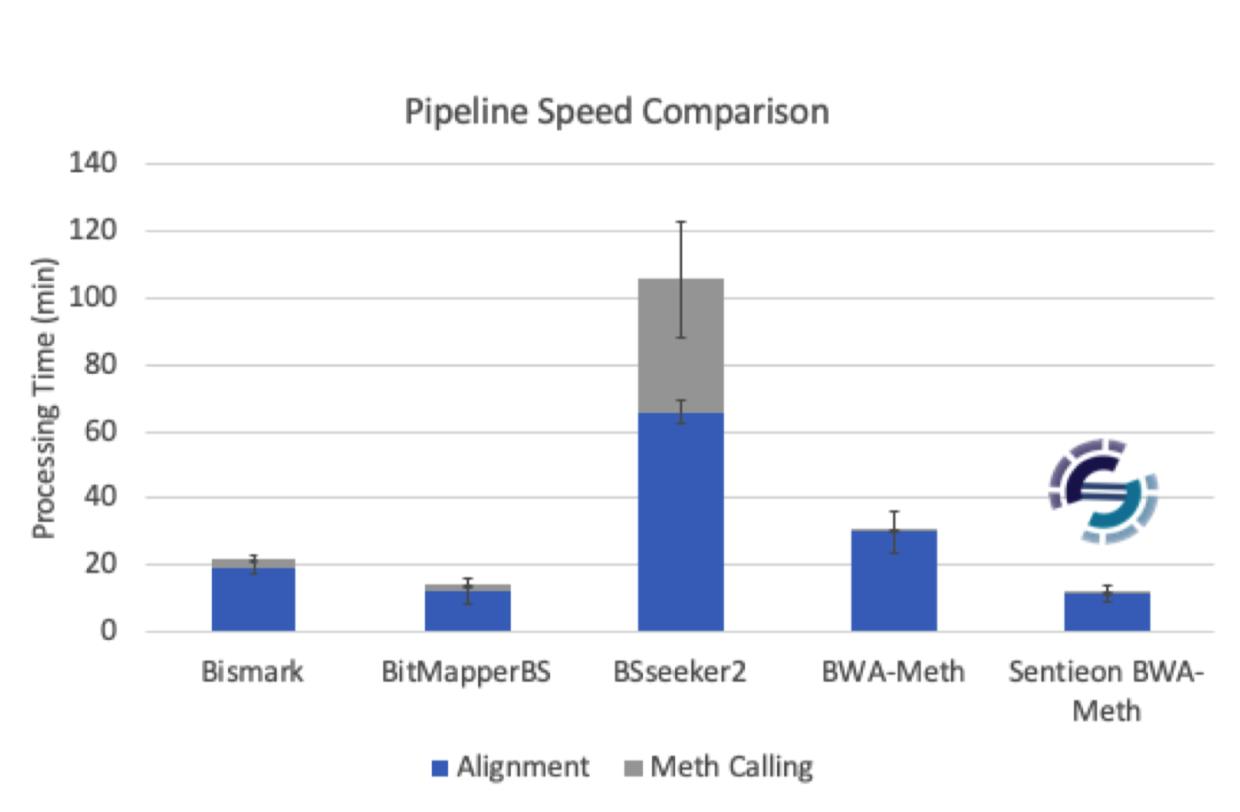
- 每次比较中,使用相同的随机种子对一百万对读取进行随机抽样。
- 在24个CPU线程的服务器上使用各软件的默认参数运行比对。
- 记录每个重复实验的性能时间。
- Sentieon BWA-Meth比原始的BWA-Meth速度提高了2.5倍,与BitMaperBS的速度相似。
-
甲基化映射准确性比较:

- 比较不同甲基组文库制备中的流程映射准确性;使用Samtools stats和Qualimap生成后比对统计数据。
- 显示库总读取的参考映射结果分布。
- Sentieon BWA-meth具有最高的主要映射率和最低的未映射率。
-
CpG位点读取覆盖率比较:

- 计算14个库和4个分析流程中识别出的CpG位点的测序覆盖率。
- 与其他测试工具相比,Sentieon BWA-Meth在CpG位点提供了更高的测序覆盖率。
-
Sentieon甲基化分析流程结论:
- Sentieon BWA-Meth与BWA-Meth提供相同的结果。
- Sentieon BWA-Meth流程显示出最高的处理速度,比开源流程快约2倍。
- Sentieon BWA-Meth具有最高的主要映射率和最高的CpG位点读取覆盖率。
-
应用说明 - 安装
- 安装bwa-meth
# Prerequisites: samtools # these 4 lines are only needed if you don\'t have toolshed installed
wget https://pypi.python.org/packages/source/t/toolshed/toolshed-0.4.0.tar.gz
tar xzvf toolshed-0.4.0.tar.gz
cd toolshed-0.4.0 sudo
python setup.py install
wget https://github.com/brentp/bwa-meth/archive/master.zip
unzip master.zip
cd bwa-meth-master
sudo python setup.py install
- 安装MethylDackel
# Prerequisites: htslib and libBigWig
git clone https://github.com/dpryan79/MethylDackel.git
cd MethylDackel
make LIBBIGWIG="/some/path/to/libBigWig.a"
make install prefix=/some/installation/path
- 安装BWA(开源)
# Only used for indexing reference genome.
git clone https://github.com/lh3/bwa.git
cd bwa; make
-
准备测试数据
- 从bwa-meth下载测试数据并使用开源BWA对参考基因组进行索引。
wget https://github.com/brentp/bwa-meth/raw/master/example/ref.fa
wget https://github.com/brentp/bwa-meth/raw/master/example/t_R1.fastq.gz
wget https://github.com/brentp/bwa-meth/raw/master/example/t_R2.fastq.gz
- 使用开源BWA构建index索引
bwameth.py index $REF #Indexes with BWA-MEM (default)
- 确保安装了开源的BWA在$PATH下,而不是Sentieon BWA

-
读取比对:
-
使用Sentieon BWA进行读取比对。
-
将Sentieon bin文件夹添加到$PATH。
-
export PATH=<PATH_TO_SENTIEON>/sentieon-genomics-202112.05/bin:$PATH
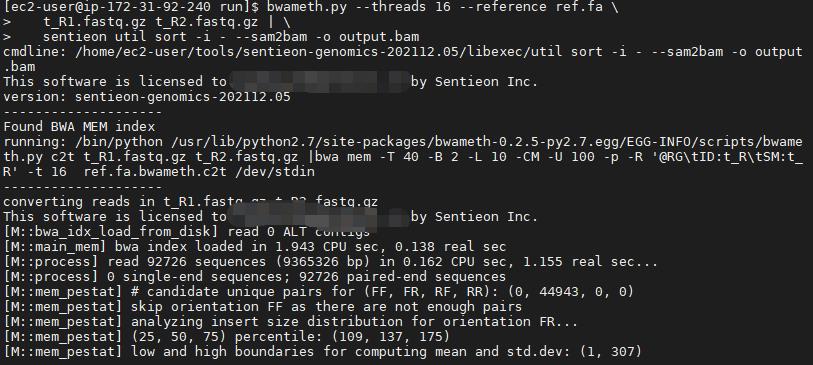
* 运行bwa-meth,通过sentieon util sort进行排序。
bwameth.py --threads 16 \\
--reference $REFERENCE \\
$FQ1 $FQ2 | \\
sentieon util sort -i - –sam2bam –o output.bam
- 确保屏幕输出以下内容。否则,bwa-meth将使用开源bwa。
This software is licensed to [xxxxx@xxxx.xxx] by Sentieon Inc.
version: sentieon-genomics-202112.06
-
制表:
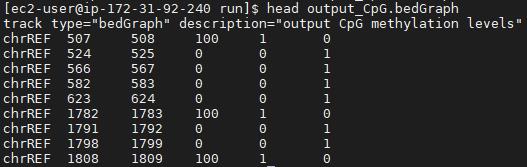
- 使用开源工具MethylDackel。
MethylDackel extract ref.fa output.bam

- 输出结果为bedGraph文件,第4列表示在给定位置有甲基化C的证据的读取/读取对数,第5列表示未甲基化C的等价值。
甲基化数据QC:使用甲基化数据计算样本间的相关性
样本间的相关性,可以反映公司加样时是否存在重复加样的错误。
下面简要介绍一下如果利用甲基化数据计算样本间的相关性
1、提取甲基化探针的snp位点、CpG的beta值
下面用的示例文件是minfi包自带的。
如果是自己的数据,那么提取甲基化snp位点用的是没有经过过滤的原始数据。
首先,安装:
BiocManager::install(c("minfi","minfiData","sva"))
library(minfi)
library(minfiData)
library(sva)
baseDir <- system.file("extdata", package="minfiData")
targets <- read.metharray.sheet(baseDir)
RGSet <- read.metharray.exp(targets = targets)
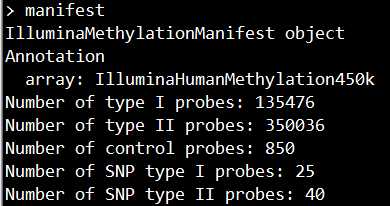
manifest <- getManifest(RGSet)
这里可以看到不同探针的情况:
一条龙服务,提取甲基化探针的snp位点、CpG的beta值:
MSet <- preprocessRaw(RGSet)
RSet <- ratioConvert(MSet, what = "both", keepCN = TRUE)
GRset <- mapToGenome(RSet)
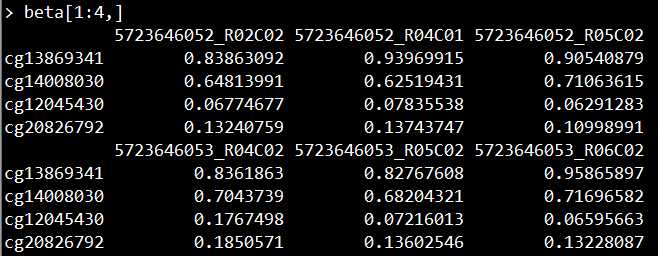
beta <- getBeta(GRset) #提取CpG的beta值
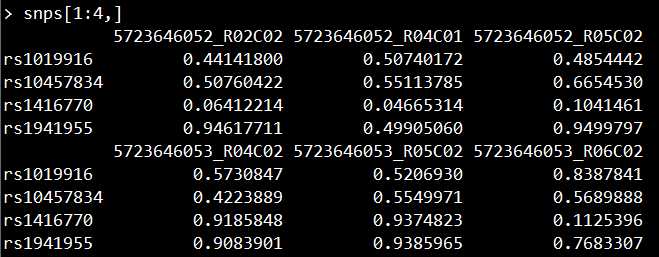
snps <- getSnpBeta(RGSet) #提取SNP位点
2、CpG和SNP的beta值位点示例结果
提取完CpG和SNP后,看一下各自的示例结果:
CpG的beta值示例结果:
甲基化SNP位点的示例结果:
3、计算相关性
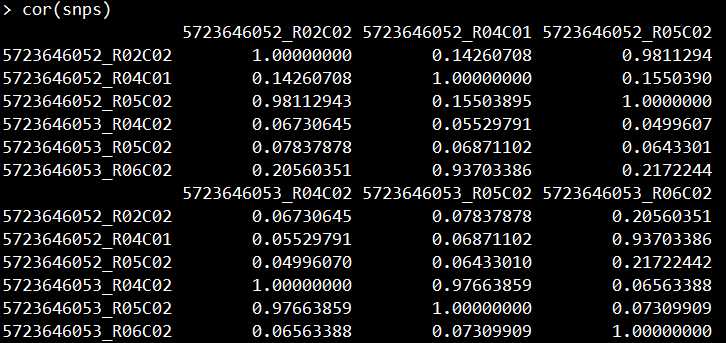
计算样本间的相关性,我们用R自带的cor函数即可。选用的数值为SNP的甲基化数值
计算相关性代码:cor(snps)
结果如下:
这里解释一下,为什么不选用CpG的beta值计算相关性。
如下图所示,我分别用了前100、1000、10000个CpG的beta值计算样本1(5723646052_R02C02)和样本2(5723646052_R04C01)的相关性,相关性均在0.97以上(蓝色框框),用snps位点计算相关性时,样本1和样本2的相关性则为0.1426071(红色框框)。
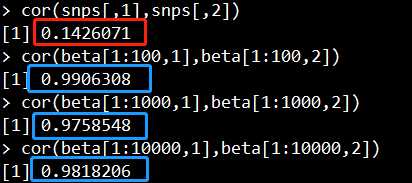
可见,CpG的beta值计算出来的相关性都特别高,根本不能区别样本间真实的相关性。
因此,计算样本间相关性,推荐甲基化探针的SNP位点。

以上是关于使用Sentieon加速甲基化(WGBS)分析的主要内容,如果未能解决你的问题,请参考以下文章
Uanle TCGA数据挖掘——预后相关的甲基化位点及构建重要基因的风险模型
r 在甲基化年龄分析的背景下,展示拦截术语如何从抽样方法中调整估计偏差