七步走纯R代码通过数据挖掘复现一篇实验文章(第1到6步)
Posted 生信技能树
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了七步走纯R代码通过数据挖掘复现一篇实验文章(第1到6步)相关的知识,希望对你有一定的参考价值。
最近给新的学徒布置了去年学徒的数据挖掘任务,让我意外的是一个影像学专业大三学生表现最好,两天就表现优异的完成了作业,而且项目代码组织习惯也非常棒,任务拆解为7个步骤,条理清晰,逻辑流畅,值得分享!
如果你也想向优秀的小伙伴看齐,欢迎加入生信技能树超级VIP计划(),如果你时间有限,也可以考虑参与我们的全国巡讲:
下面看学徒表演
本期我们的任务是全代码复现一篇paper,标题为 :Co-expression networks revealed potential core lncRNAs in the triple-negative breast cancer. PMID:27380926 (大家需要自行阅读文献才明白jimmy交给学徒的任务!)文章里面是自己测了8个TNBC病人的转录组然后分析,但是我们有TCGA数据库,所以可以复现,这就是为什么标题是七步走纯R代码通过数据挖掘复现一篇实验文章!
主要流程:
下载数据
数据清洗
质量控制
差异分析
注释mRNA,lncRNA
富集分析
WGCNA(因为排版限制,内容见本期第3条推文)
Step1. 数据下载
这里参考去年的学徒数据挖掘:
表达矩阵下载及临床信息下载
GTF文件
Step2. 数据清洗
提高数据清洗的能力,将会很大程度的提高你做分析数据的速度,可能有的人还是习惯用Excel来清洗数据,但是我建议能用代码的尽量用代码解决,数据清洗思路也很重要,一定要清楚你的目标,然后思考可能实现的途径。
首先提取出三阴性乳腺癌样本,这里参考去年的学徒数据挖掘: 可以看到是118个TNBC病人
# 挑选三阴性乳腺癌的样本
### FALSE TRUE
## 1165 118
tnbc_sample = phenotype_file[phenotype_file$breast_carcinoma_estrogen_receptor_status == 'Negative' &
phenotype_file$breast_carcinoma_progesterone_receptor_status == 'Negative' &
phenotype_file$lab_proc_her2_neu_immunohistochemistry_receptor_status == 'Negative', ]
save(tnbc_sample,file = '../02_data/tnbc_sample.Rdata')
然后在118个三阴性乳腺癌中找到有N-T配对样本的病人
在TCGA中第14,15位的数字01~09代表肿瘤样本,10以上则为正常样本
# 把肿瘤样本提取出来,把正常样本提取出来然后根据前十二字符merged到的样本就是属于配对样本
library(stringr)
b = tnbc_sample[1:2]
tumor_sample <- b[substr(b$submitter_id.samples,14,15) < 10,]
tumor_sample$TCGA_ID <- str_sub(tumor_sample$submitter_id.samples, 1, 12)
normal_sample <- b[!substr(b$submitter_id.samples,14,15) < 10,]
normal_sample$TCGA_ID <- str_sub(normal_sample$submitter_id.samples, 1, 12)
tmp = merge(normal_sample, tumor_sample, by = "TCGA_ID")
# 以下是为了方便后续提取数据
a = tmp[,2:3]
colnames(a)
## [1] "submitter_id.samples.x"
## [2] "additional_pharmaceutical_therapy.x"
names(a) = c("submitter_id.samples", "additional_pharmaceutical_therapy")
b = tmp[,4:5]
names(b) = c("submitter_id.samples", "additional_pharmaceutical_therapy")
TNBC_pair_sample = rbind(a,b)
head(TNBC_pair_sample)
save(TNBC_pair_sample, file = "../02_data/TNBC_pair_sample.Rdata")
在配对样本中过滤掉并非同时有正常和肿瘤组织测序的样本
rm(list = ls())
library(readr)
# 提取表达矩阵
load(file = "../02_data/TNBC_pair_sample.Rdata")
rawdata <- read_tsv("../02_data/TCGA-BRCA.htseq_counts.tsv")
rawdata[1:3,1:3]
## # A tibble: 3 x 3
## Ensembl_ID `TCGA-3C-AAAU-01A` `TCGA-3C-AALI-01A`
## <chr> <dbl> <dbl>
## 1 ENSG00000000003.13 9.35 8.71
## 2 ENSG00000000005.5 1.58 1.58
## 3 ENSG00000000419.11 10.9 10.8
rawdata = as.data.frame(rawdata)
rownames(rawdata) = rawdata[,1]
rawdata = rawdata[, -1]
rawdata[1:3,1:3]
## TCGA-3C-AAAU-01A TCGA-3C-AALI-01A TCGA-3C-AALJ-01A
## ENSG00000000003.13 9.348728 8.714246 10.35645
## ENSG00000000005.5 1.584963 1.584963 5.72792
## ENSG00000000419.11 10.874981 10.834471 10.32980
table(colnames(rawdata) %in% TNBC_pair_sample$submitter_id.samples)
##
## FALSE TRUE
## 1191 26
expr = rawdata[,colnames(rawdata) %in% TNBC_pair_sample$submitter_id.samples]
expr = as.data.frame(t(expr))
expr[1:3,1:3]
## ENSG00000000003.13 ENSG00000000005.5 ENSG00000000419.11
## TCGA-A7-A4SE-01A 10.89482 4.906891 10.903882
## TCGA-AC-A2BK-01A 13.06811 0.000000 11.758640
## TCGA-AC-A2QJ-01A 10.27496 4.643856 9.763212
# 还要继续过滤掉不符合要求的样本
b = expr[1:2]
b$submitter_id.samples = rownames(b)
# 为了去掉其中一个样本测了两次tumor
tumor_sample <- b[substr(b$submitter_id.samples,14,16) == "01A",]
tumor_sample$TCGA_ID <- str_sub(tumor_sample$submitter_id.samples, 1, 12)
normal_sample <- b[!substr(b$submitter_id.samples,14,15) < 10,]
normal_sample$TCGA_ID <- str_sub(normal_sample$submitter_id.samples, 1, 12)
tmp = merge(normal_sample, tumor_sample, by = "TCGA_ID")
###-----------------------------------------------------------------------------
a = tmp[,2:4]
colnames(a)
## [1] "ENSG00000000003.13.x" "ENSG00000000005.5.x"
## [3] "submitter_id.samples.x"
names(a) = c("ENSG00000000003.13", "ENSG00000000005.5", "submitter_id.samples")
b = tmp[,5:7]
names(b) = c("ENSG00000000003.13", "ENSG00000000005.5", "submitter_id.samples")
TNBC_pair_sample = rbind(a,b)
table(table(str_sub(TNBC_pair_sample$submitter_id.samples,1,12)))
##
## 2
## 10
# ---------------------------------------------------------------------------------
table(rownames(expr) %in% TNBC_pair_sample$submitter_id.samples)
##
## FALSE TRUE
## 6 20
expr = expr[rownames(expr) %in% TNBC_pair_sample$submitter_id.samples,]
dim(expr)
## [1] 20 60483
save(expr,file = "../02_data/TNBC_pair_expr.Rdata")
Step3. 质量控制
表达矩阵质量控制还有很多其他的图和方式,例如PCA等等,需要大家了解
rm(list = ls())
load(file = "../02_data/TNBC_pair_expr.Rdata")plot(hclust(dist(expr)))

rownames(expr)# 学徒在这里考虑到模仿的文章里面是8个病人,所以这里剔除了2个病人。# 考虑的是hclust的离群点
exprSet = expr[c(-5,-6,-13,-14),]
save(exprSet,file = "../02_data/TCGA_TNBC_QC_exprSet.Rdata")
step4. 差异分析
差异分析可选edger,limma,DESeq2,这里使用DESeq2
rm(list = ls())
library(stringr)
library(DESeq2)
library(DT)
load(file = "../02_data/TCGA_TNBC_QC_exprSet.Rdata")
raw_data = exprSet
raw_data <- as.data.frame( t(raw_data) )
raw_data[1:5, 1:6]
## TCGA-BH-A0B3-01A TCGA-BH-A0B3-11B TCGA-BH-A18V-01A
## ENSG00000000003.13 11.287135 12.728346 10.100662
## ENSG00000000005.5 3.584963 8.864186 4.643856
raw_data <- 2^raw_data - 1
raw_data <- ceiling( raw_data )
raw_data[1:5, 1:6]
## TCGA-BH-A0B3-01A TCGA-BH-A0B3-11B TCGA-BH-A18V-01A
## ENSG00000000003.13 2499 6785 1097
## ENSG00000000005.5 11 465 24
pick_row <- apply( raw_data, 1, function(x){
sum(x == 0) < 16
})
raw_data <- raw_data[pick_row, ]
dim(raw_data )
## [1] 48236 16
# 使用DESeq2需要表达矩阵和group_list
a = as.data.frame(rownames(exprSet))
names(a) = "sample_id"
group_list=factor(ifelse(as.numeric(substr(colnames(raw_data),14,15)) < 10,'tumor','normal'))
table(group_list)
## group_list
## normal tumor
## 8 8
colData <- data.frame(row.names=colnames(raw_data),
group_list=group_list)
## 第一列有名称时,tidy=TRUE
raw_data[1:3,1:3]
## TCGA-BH-A0B3-01A TCGA-BH-A0B3-11B TCGA-BH-A18V-01A
## ENSG00000000003.13 2499 6785 1097
## ENSG00000000005.5 11 465 24
## ENSG00000000419.11 1853 1921 3564
head(colData)
## group_list
## TCGA-BH-A0B3-01A tumor
## TCGA-BH-A0B3-11B normal
dds <-DESeqDataSetFromMatrix(countData=raw_data,
colData=colData,
design=~group_list,
tidy=FALSE)
dds <- DESeq(dds)
vsd <- vst(dds, blind = FALSE)
# 制作表达矩阵小心结果相反,这里是tumor vs. normal
contrast <- c("group_list","tumor","normal")
dd1 <- results(dds, contrast=contrast, alpha = 0.05)
plotMA(dd1, ylim=c(-2,2))

# lfcShrink矫正
dd2 <- lfcShrink(dds, contrast=contrast, res=dd1)
plotMA(dd2, ylim=c(-2,2))

summary(dd2, alpha = 0.05)
##
## out of 48212 with nonzero total read count
## adjusted p-value < 0.05
## LFC > 0 (up) : 4968, 10%
## LFC < 0 (down) : 4310, 8.9%
## outliers [1] : 0, 0%
## low counts [2] : 14979, 31%
## (mean count < 1)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?results
library(dplyr)
library(tibble)
res <- dd2 %>%
data.frame() %>%
rownames_to_column("gene_id")
DT::datatable(res)
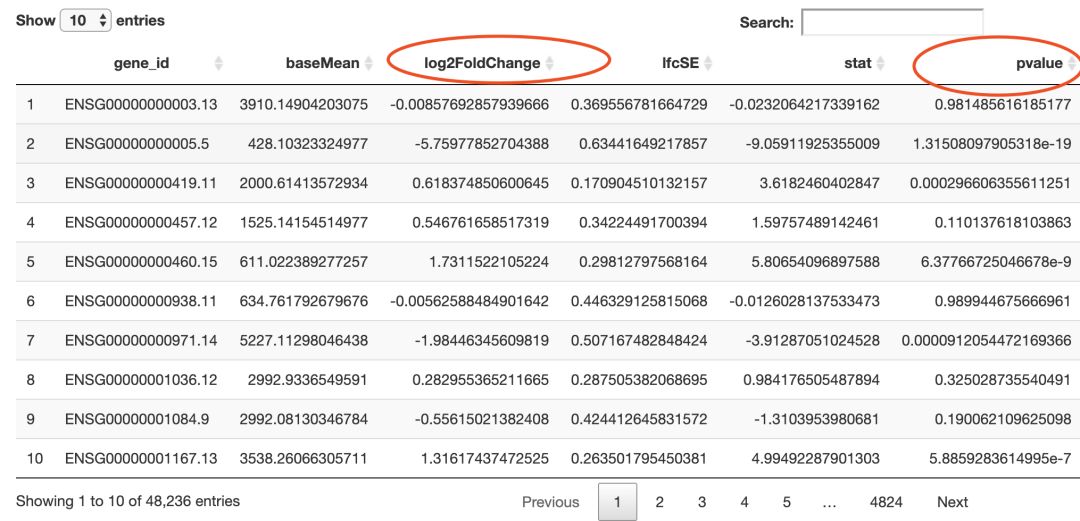
save(res,vsd, file = "../02_data/TCGA_TNBC_different_analysis.Rdata")
step5. 基因注释
lncRNA注释还是有挺多方法的,gencode上的文件同样可以可以来注释,至于哪些类型的注释是属于lncRNA需要自己查看相关的资料
# 加载包
# -------------------------------------------------------------------------------
rm(list = ls())
Sys.setenv(LANGUAGE = "en") #显示英文报错信息
options(stringsAsFactors = FALSE) #禁止chr转成factor
library(rtracklayer)
library(dplyr)
library(tidyr)
library(pheatmap)
# 构建基因注释的GTF文件
# --------------------------------------------------------------------------------
gtf1 <- rtracklayer::import('../02_data/Homo_sapiens.GRCh38.97.chr.gtf')
gtf_df <- as.data.frame(gtf1)
colnames(gtf_df)
## [1] "seqnames" "start"
## [3] "end" "width"
## [5] "strand" "source"
## [7] "type" "score"
## [9] "phase" "gene_id"
## [11] "gene_version" "gene_name"
## [13] "gene_source" "gene_biotype"
gtf = gtf_df[,c(10,14)]
head(gtf)
## gene_id gene_biotype
## 1 ENSG00000223972 transcribed_unprocessed_pseudogene
## 2 ENSG00000223972 transcribed_unprocessed_pseudogenesave(gtf,file = "../02_data/Homo_sapiens.GRCh38.97.chr.Rdata")
# 注释mRNA并找出显著差异的mRNA# ----------------------------------------------------------------------------------
load(file = "../02_data/TCGA_TNBC_different_analysis.Rdata")
res_nopoint <- res %>%
tidyr::separate(gene_id,into = c("gene_id"),sep="\\.")
res_nopoint[1:3,1:3]## gene_id baseMean log2FoldChange## 1 ENSG00000000003 3910.1490 -0.008576929## 2 ENSG00000000005 428.1032 -5.759778527## 3 ENSG00000000419 2000.6141 0.618374851mRNA_exprSet <- gtf_df %>%
dplyr::filter(type=="gene",gene_biotype=="protein_coding") %>% #筛选gene,和编码指标
dplyr::select(c(gene_name,gene_id,gene_biotype)) %>%
dplyr::inner_join(res_nopoint,by ="gene_id") %>%
tidyr::unite(gene_id,gene_name,gene_id,gene_biotype,sep = "|")
dim(mRNA_exprSet)## [1] 18949 7
diffSig_mRNA <- mRNA_exprSet[with(mRNA_exprSet, (abs(log2FoldChange)>2 & padj < 0.05 )), ]
dim(diffSig_mRNA)## [1] 2308 7# 注释lncRNA并找出显著差异的lncRNA# ----------------------------------------------------------------------------------
table(gtf_df$gene_biotype)LncRNA_exprSet <- gtf_df %>%
dplyr::filter(type=="gene",gene_biotype %in% "lncRNA") %>% #注意这里是transcript_biotype
dplyr::select(c(gene_name,gene_id,gene_biotype)) %>%
dplyr::distinct() %>% #删除多余行
dplyr::inner_join(res_nopoint,by ="gene_id") %>%
tidyr::unite(gene_id,gene_name,gene_id,gene_biotype,sep = "|")dim(LncRNA_exprSet)## [1] 12065 7
diffSig_lncRNA <- LncRNA_exprSet[with(LncRNA_exprSet, (abs(log2FoldChange)>2 & padj < 0.05 )), ]
dim(diffSig_lncRNA)## [1] 1127 7# 提取显著差异的mRNA,lncRNA表达矩阵,并进行ID转换# -----------------------------------------------------------------------
vst = as.data.frame(assay(vsd))vst$gene_id = rownames(vst)
vst_nopoint <- vst %>%
tidyr::separate(gene_id,into = c("gene_id"),sep="\\.")
vst_nopoint[1:3,1:3]rownames(vst_nopoint) = vst_nopoint$gene_id
vst_nopoint[1:3,1:3]vst_nopoint = vst_nopoint[, -ncol(vst_nopoint)]
vst_nopoint[1:3,1:3]# 构建lncRNA_ids#-----------------------------------------------------------------------------------------
a = as.data.frame(diffSig_lncRNA$gene_id %>%
str_split("\\|",simplify = T))
names(a) = c("symbol", "probe_id","gene_type")
# diffSig_lncRNA ID转换#-----------------------------------------------------------------------------------------
ids = a[,1:2]
exprSet_lncRNA = vst_nopoint
exprSet_lncRNA[1:3,1:3] class(ids$symbol)## [1] "character"
table(rownames(exprSet_lncRNA) %in% ids$probe_id)## ## FALSE TRUE ## 47197 1039
exprSet_lncRNA = exprSet_lncRNA[rownames(exprSet_lncRNA) %in% ids$probe_id,]
dim(exprSet_lncRNA)## [1] 1039 16
ids = ids[match(rownames(exprSet_lncRNA), ids$probe_id),]
tmp = by(exprSet_lncRNA,
ids$symbol,
function(x) rownames(x)[which.max(rowMeans(x)
)])
probes = as.character(tmp)
dim(tmp)## [1] 1039
exprSet_lncRNA = exprSet_lncRNA[rownames(exprSet_lncRNA) %in% probes, ]
dim(exprSet_lncRNA)## [1] 1039 16
rownames(exprSet_lncRNA)=ids[match(rownames(exprSet_lncRNA),ids$probe_id),1]
exprSet_lncRNA[1:3,1:3]# 构建mRNA_ids#----------------------------------------------------------------------------------------
a = as.data.frame(diffSig_mRNA$gene_id %>%
str_split("\\|",simplify = T))
names(a) = c("symbol", "probe_id","gene_type")
# diffSig_mRNA ID转换# --------------------------------------------------------------------------------------
ids = a[,1:2]
exprSet_mRNA = vst_nopointexprSet_mRNA = exprSet_mRNA[rownames(exprSet_mRNA) %in% ids$probe_id,]
dim(exprSet_mRNA)## [1] 2273 16
ids = ids[match(rownames(exprSet_mRNA), ids$probe_id),]tmp = by(exprSet_mRNA,
ids$symbol,
function(x) rownames(x)[which.max(rowMeans(x)
)])
probes = as.character(tmp)
dim(tmp)## [1] 2272
exprSet_mRNA = exprSet_mRNA[rownames(exprSet_mRNA) %in% probes, ]
exprSet_mRNA[1:3,1:3]## TCGA-BH-A0B3-01A TCGA-BH-A0B3-11B TCGA-BH-A18V-01A## ENSG00000000005 4.443206 8.357049 5.006992## ENSG00000003249 11.148636 8.672442 12.093123## ENSG00000003436 8.780552 12.019202 10.205594
dim(exprSet_mRNA)## [1] 2272 16
rownames(exprSet_mRNA)=ids[match(rownames(exprSet_mRNA),ids$probe_id),1]
exprSet_mRNA[1:3,1:3]## TCGA-BH-A0B3-01A TCGA-BH-A0B3-11B TCGA-BH-A18V-01A## TNMD 4.443206 8.357049 5.006992## DBNDD1 11.148636 8.672442 12.093123## TFPI 8.780552 12.019202 10.205594
画个显著差异mRNA的热图和火山图
# diffSig_mRNA热图
#--------------------------------------------------------------------------------
group_list=factor(ifelse(as.numeric(substr(colnames(exprSet_lncRNA),14,15)) < 10,'tumor','normal'))
table(group_list)
(colData <- data.frame(row.names=colnames(exprSet_lncRNA),
group_list=group_list))
pheatmap(exprSet_lncRNA,scale = "row",show_rownames = FALSE, show_colnames = FALSE,
annotation_col = colData)

## mRNA火山图
# -----------------------------------------------------------------------------------
logFC_cutoff = 2
DEG = mRNA_exprSet
DEG = na.omit(DEG)
DEG$change = as.factor( ifelse( DEG$padj< 0.05 & abs(DEG$log2FoldChange) > logFC_cutoff,
ifelse( DEG$log2FoldChange > logFC_cutoff , 'UP', 'DOWN' ), 'NOT' ) )
table(DEG$change)
##
## DOWN NOT UP
## 1228 15548 1045
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(DEG[DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$change =='DOWN',])
)
library(ggplot2)
g = ggplot(data=DEG,
aes(x=log2FoldChange, y=-log10(padj),
color=change)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red')) ## corresponding to the levels(res$change)
print(g)

lncRNA同样可以画出相应的图
# diffSig_lncRNA热图
# ----------------------------------------------------------------------------------------
group_list=factor(ifelse(as.numeric(substr(colnames(exprSet_mRNA),14,15)) < 10,'tumor','normal'))
table(group_list)
## group_list
## normal tumor
## 8 8
(colData <- data.frame(row.names=colnames(exprSet_mRNA),
group_list=group_list))
## group_list
## TCGA-BH-A0B3-01A tumor
## TCGA-BH-A0B3-11B normal
## TCGA-BH-A18V-01A tumor
## TCGA-BH-A18V-11A normal
## TCGA-BH-A1F6-01A tumor
## TCGA-BH-A1F6-11B normal
## TCGA-BH-A1FC-01A tumor
## TCGA-BH-A1FC-11A normal
## TCGA-E2-A158-01A tumor
## TCGA-E2-A158-11A normal
## TCGA-E2-A1LH-01A tumor
## TCGA-E2-A1LH-11A normal
## TCGA-E2-A1LS-01A tumor
## TCGA-E2-A1LS-11A normal
## TCGA-GI-A2C9-01A tumor
## TCGA-GI-A2C9-11A normal
pheatmap(exprSet_mRNA,scale = "row",show_rownames = FALSE, show_colnames = FALSE,
annotation_col = colData)

# lncRNA 火山图
# -------------------------------------------------------------------------------------
logFC_cutoff = 2
DEG = LncRNA_exprSet
DEG = na.omit(DEG)
DEG$change = as.factor( ifelse( DEG$padj< 0.05 & abs(DEG$log2FoldChange) > logFC_cutoff,
ifelse( DEG$log2FoldChange > logFC_cutoff , 'UP', 'DOWN' ), 'NOT' ) )
table(DEG$change)
##
## DOWN NOT UP
## 611 7067 428
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(DEG[DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$change =='DOWN',])
)
library(ggplot2)
g = ggplot(data=DEG,
aes(x=log2FoldChange, y=-log10(padj),
color=change)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red')) ## corresponding to the levels(res$change)
print(g)

save(mRNA_exprSet,LncRNA_exprSet,diffSig_lncRNA, diffSig_mRNA, exprSet_mRNA,exprSet_lncRNA,
file = "../02_data/TCGA_TNBC_pair_annotation_result.Rdata")
step6.富集分析
rm(list = ls())
library("clusterProfiler")
library("org.Hs.eg.db")
library(ggplot2)
library(RColorBrewer)
library(gridExtra)
library(enrichplot)
#library(plyr)
library(ggrepel)
library(stringr)
load(file = "../02_data/TCGA_TNBC_pair_annotation_result.Rdata")
mRNA = as.data.frame(diffSig_mRNA$gene_id %>%
str_split("\\|",simplify = T))
names(mRNA) = c("symbol", "probe_id","gene_type")
mRNA = na.omit(mRNA)
gene.df <- bitr(mRNA$symbol, fromType="SYMBOL",
toType="ENTREZID",
OrgDb = "org.Hs.eg.db")
ego_BP <- enrichGO(gene = gene.df$ENTREZID,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "BP",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE)
dotplot(ego_BP, showCategory = 20,font.size = 8)

# ?dotplot
ego_CC <- enrichGO(gene = gene.df$ENTREZID,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "CC",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE)
dotplot(ego_CC, showCategory = 20,font.size = 8)

ego_MF <- enrichGO(gene = gene.df$ENTREZID,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "MF",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE)
dotplot(ego_MF, showCategory = 20,font.size = 8)

ego_KEGG <- enrichKEGG(gene = gene.df$ENTREZID, organism = "hsa",
keyType = "kegg",
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
minGSSize = 10, maxGSSize = 500,
qvalueCutoff = 0.05,
use_internal_data = FALSE)
dotplot(ego_KEGG, showCategory = 20,font.size = 8)

save(ego_BP, ego_CC,ego_MF,ego_KEGG, file = "../02_data/TCGA_TNBC_pair_enrichment.Rdata")
富集分析本身的难度不大,但是如何利用富集到的通路去讲你的生物学故事,则十分重要
因为内容太多,微信推文排版限制,第七步WGCNA需要大家移步到本期第3条查看,谢谢!
另外,本文是rmarkdown格式,如果需要文章pdf以及rmarkdown源代码和网页报告,需要后台回复:优秀学徒,即可获取!
全国巡讲约你
(已结束)
(已结束)
以上是关于七步走纯R代码通过数据挖掘复现一篇实验文章(第1到6步)的主要内容,如果未能解决你的问题,请参考以下文章
PHPOK任意文件上传漏洞复现(CVE-2018-8944)