GEO数据挖掘流程+STRING VS R in KEGG/GO
Posted 生信技能树
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GEO数据挖掘流程+STRING VS R in KEGG/GO相关的知识,希望对你有一定的参考价值。
In molecular biology, STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) is a biological database and web resource of known and predicted protein–protein interactions.(from Wkkipedia)
大家好,我是在生信技能书练习了2月半的学徒,爱好是唱,跳,rap和生信。之前在复现论文的时候,有遇到过PPI网络,老师说可以使用STRING这个网页工具来完成。这个PPI网络图不仅可以表达蛋白之间的相互作用,而且还能把相应基因的功能展现出来。其实还可以做KEGG/GO注释,那么R语言的代码也可以做,他们二者的区别是什么呢?就是我们接下来要讲的东西啦。
首先,在生信学徒培训的前一个半月里,主要是攻克了R语言,然后做了一些RNA-seq和GEO数据的挖掘。这里展示一下GEO数据挖掘的流程。
下载数据(这里提供三种方式)
1.GEO主页下载原始数据(RAW.tar)
下载下来是CEL格式,需要自己进行预处理。hgu 95系列和133系列的芯片常用affy包中ReadAffy函数进行读取,有些平台用affy包处理不了,例如[HuGene-1_1-st] Affymetrix Human Gene 1.1 ST Array这个平台,就需要oligo包read.celfiles函数进行读取。illumina的芯片用lumi包来处理。之后可以进行rma或mas5进行归一化操作。
2.GEO主页下载series_matrix.txt文件
exprSet = read.table('GSE42872_series_matrix.txt.gz', sep = '\t', fill = T, comment.char = "!", header = T)
3.GEOquery包直接下载表达量
library(GEOquery)
gset = getGEO('GSE42872') #返回一个list
exprSet = exprs(gset[[1]])
getGEO这个函数,输入不同的参数,下载的东西不一样。输入参数是GDS号时,下载soft文件,参数是GPL号时,下载芯片设计信息,参数是GSE号时,下载series_matrix.txt.gz文件,返回的是ExpressionSet对象,需要掌握geneNames,sampleNames,pData,exprs等对ExpressionSet对象操作的函数。
ID转换、表达矩阵
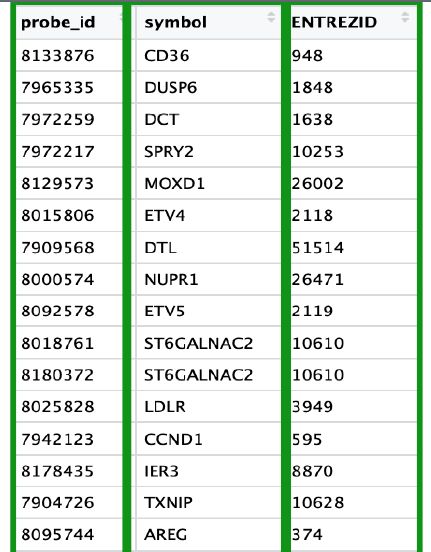
从GEO上下载的表达谱的行名是probe_id探针名,但是不同的平台,探针名不同,我们也无法直观地知道某个样本在某个探针上的表达量是那个基因的表达量,于是就需要将探针名转换为大家公认的NCBI的entrez ID,或者HUGO组织规定的gene symbol以便于后续分析。于是,我们要根据不同的GPL找到该芯片平台有对应的bioconductor注释包来找到探针与基因的对应关系,再进行转换。这里会遇到,“一个探针对应着多个基因”或者“一个基因对应多个探针”或者“探针没有对应基因”的情况,这就需要过滤整合表达矩阵,处理方法不尽相同。
表达矩阵描述的就是各个基因在各个样本上的表达量。这讲主要是表达矩阵的可视化。无论是芯片表达数据或是转录组高通量测序数据,下载完表达谱需要根据生物学背景验证一下表达谱是不是正确的。只有确定了所得表达谱是正确的,之后的差异分析等一系列分析手段才是有意义的。这里提到的方法是看看管家基因的表达量是不是在表达谱中处于高表达。也可以用boxplot看看每个样本的表达量分布图,看看是否有批次效应等等。这里就需要去了解一些画图函数的使用方法。
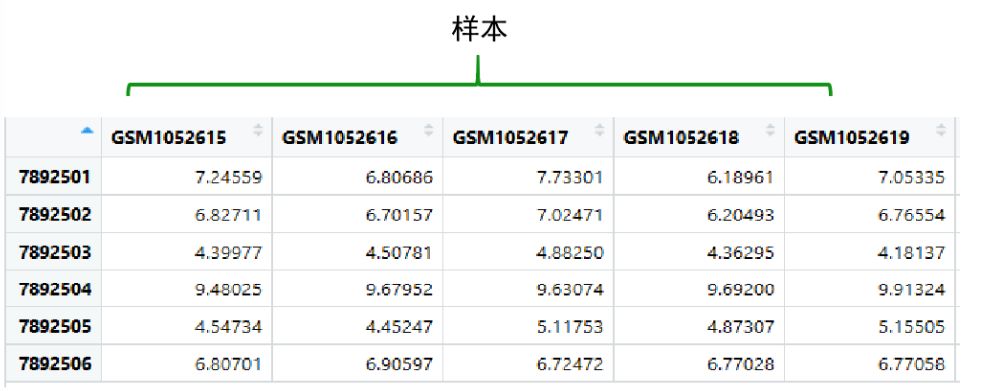
表达矩阵的提取(这里的‘GSE42872’是个例子)
library(GEOquery)
gset <- getGEO('GSE42872', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
save(gset,file='GSE42872_eSet.Rdata') ## 保存到本地
}
class(gset) #查看数据类型
length(gset)
class(gset[[1]])
gset
# assayData: 33297 features, 6 samples
# 因为这个GEO数据集只有一个GPL平台,所以下载到的是一个含有一个元素的list
a=gset[[1]]
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
分组信息的话,就通过下面的方法得到。而且,分组信息的个数和样本是一一对应的。
library(GEOquery)
gset <- getGEO('GSE42872', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
a=gset[[1]]
dat=exprs(a)
pd=pData(a) #通过查看说明书知道取对象a里的临床信息用pData
## 挑选一些感兴趣的临床表型。
library(stringr)
group_list=str_split(pd$title,' ',simplify = T)[,4]
table(group_list)
在R中看到的是这样子的:

差异分析及可视化
差异分析呢,就是把表达量特别高和表达量特别低的基因给筛选出来,因为理论上,只有这种不平凡的基因,才会对你想要研究的东西影响最大。提取出来了之后,用图形和表格直观地展示出来,就是所谓的数据可视化。
下面的代码,就是在R中,设置条件,筛选出差异基因DEGs(differentially expressed genes).一般来说,火山图,MA图和热图都是我们DEGs可视化的选择。
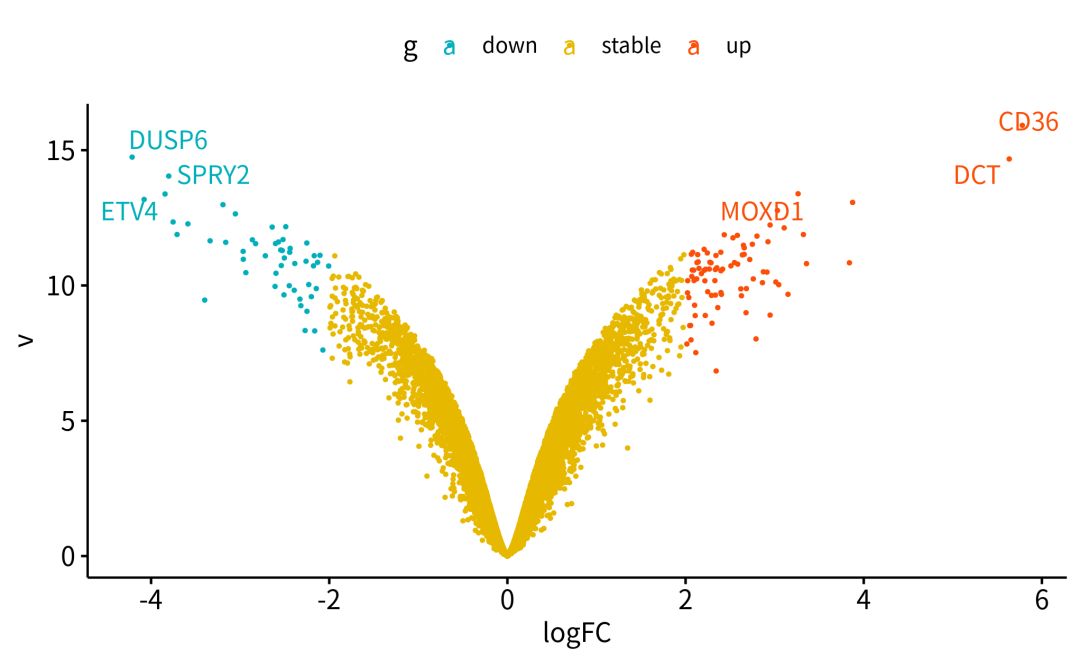
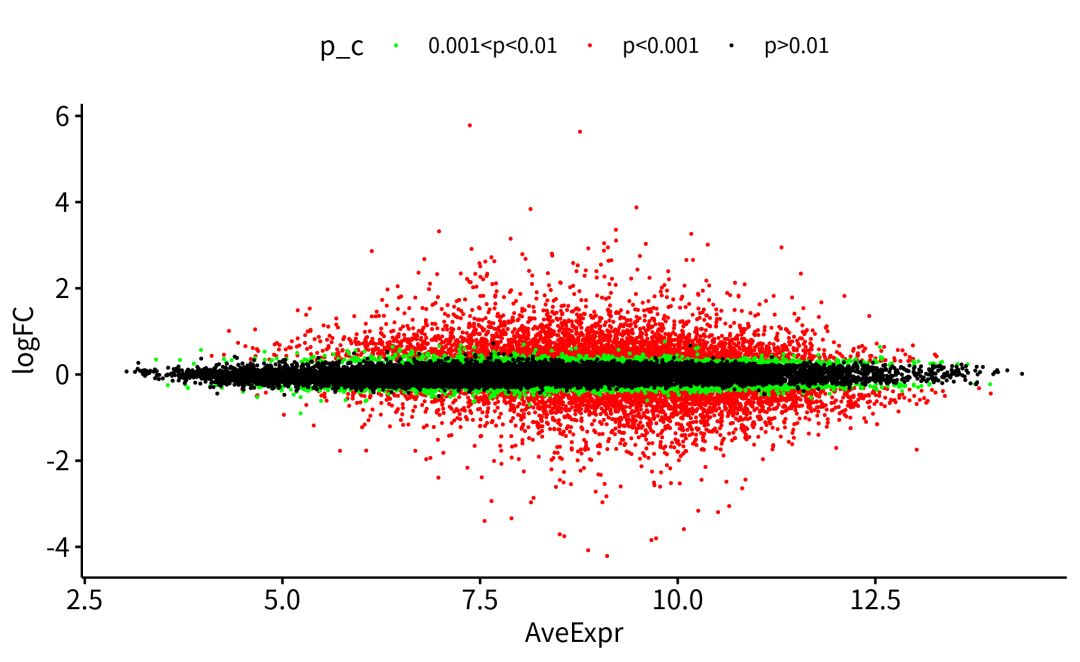
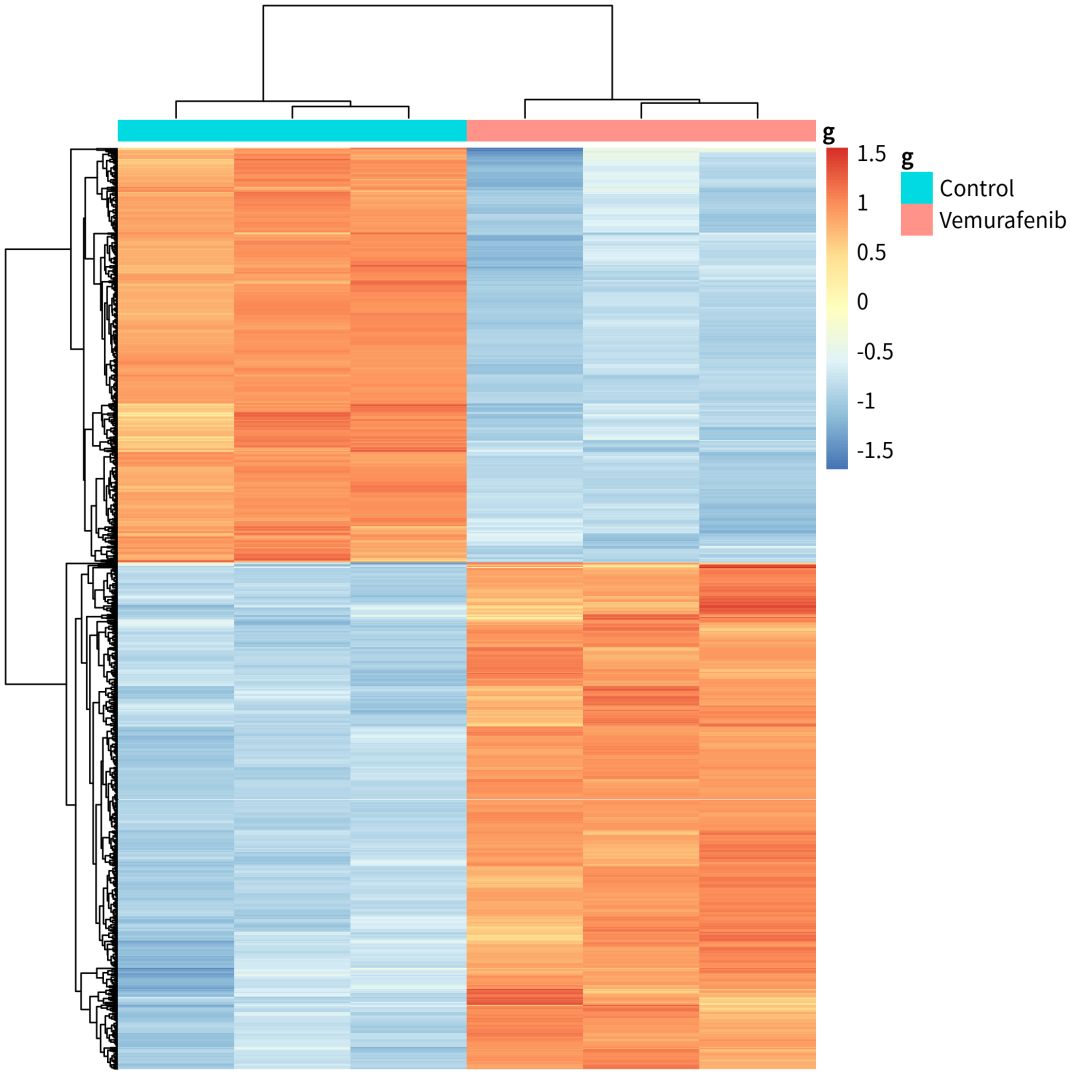
###不知道怎么作差异分析和可视化,不知道怎么用R。无所谓,大神已经把代码post到这里https://github.com/jmzeng1314/GEO,操作顺序放了在这里。https://www.bilibili.com/video/av25643438
富集分析KEGG、GO注释
介绍基因的注释及富集分析。差异分析通过自定义的阈值挑选了有统计学显著的基因列表后我们其实是需要对它们进行注释才能了解其功能,最常见的就是GO/KEGG数据库注释,当然也可以使用Reactome和Msigdb数据库来进行注释。而最常见的注释方法就是超几何分布检验。
当然还有其他的注释方法。超几何分布检验,运用到通路的富集概念就是“总共有多少基因(这个地方值得注意,主流认为只考虑那些在KEGG等数据库注释的背景基因),你的通路有多少基因,你的通路被抽中了多少基因(在差异基因里面属于你的通路的基因)。”目的就是知道,哪些通路中的哪些基因的表达因为药物或者某些操作的作用发生了较大的变化,导致通路有较大改变。
KEGG输入的基因是EntrezID,在此之前需要进行转换。当然,上面的ID转换已经包括在里面了,其实蛮多人是会嫌麻烦漏掉这一步的。
在R中如何进行注释,这里就不在多说,不知道如何运用R或者还没有试过在R中进行GO/KEGG注释的小伙伴们,可以到JM大神的b站观看视频。
https://www.bilibili.com/video/av25643438https://www.bilibili.com/video/av26731585
看完教学视频,下面的图表唾手可得!!!
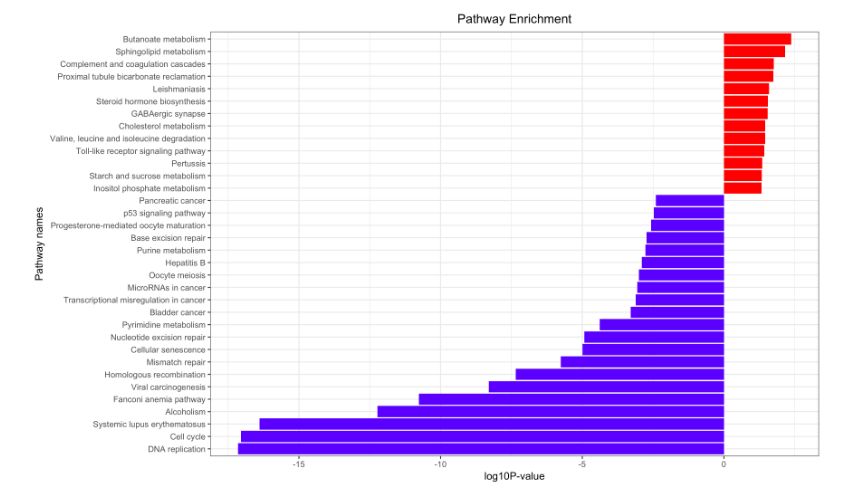
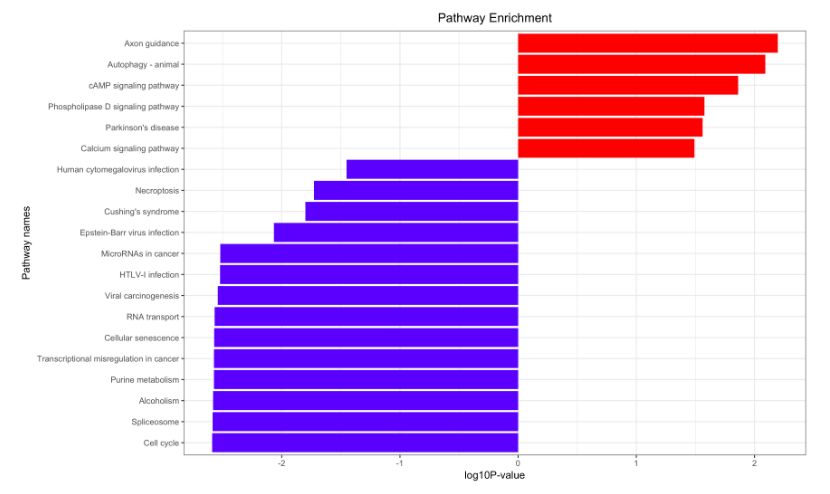
STRING的基操和文件下载
我们得到了筛选出来的DEGs,还可以通过包来做ID转换,把symbol转换成ENSEMBL的蛋白ID。但是,之前本人转换过了,发现ENSEMBL的prot ID post上去匹配不了,后来的某天早晨,由于看了cxk的篮球视频,我直接把symbol list放上了STRING,发现居然可以识别,而且自动匹配成对应的蛋白ID!
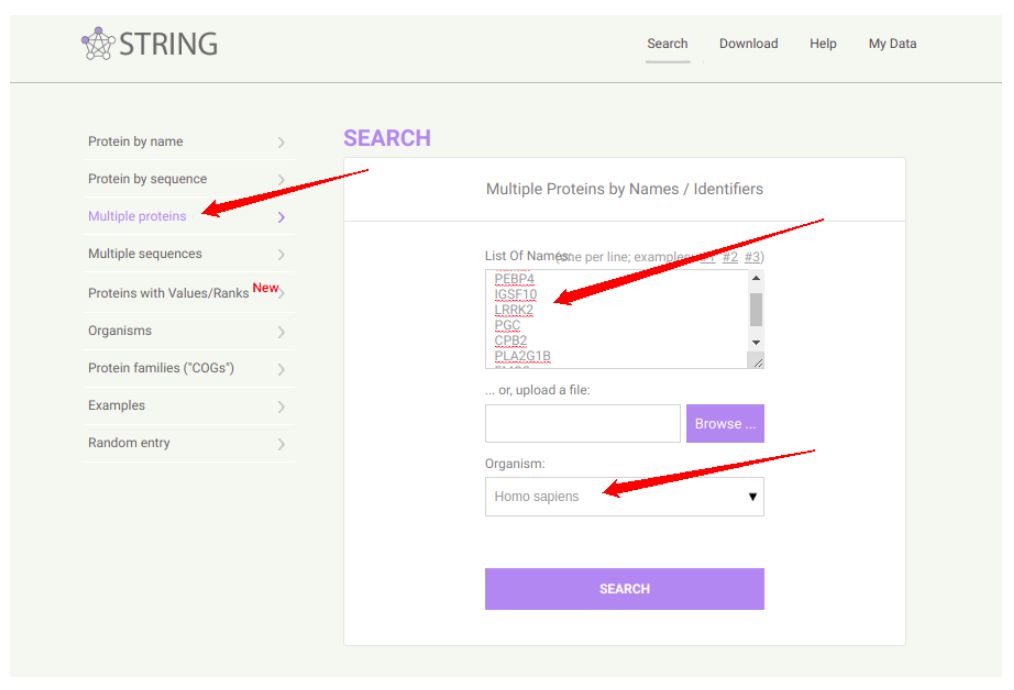
只要把你的基因粘贴到右边的大方框,下面选好物种就OK了。当然,记得左边选择第三个,除非你是有蛋白ID或者是AA序列。
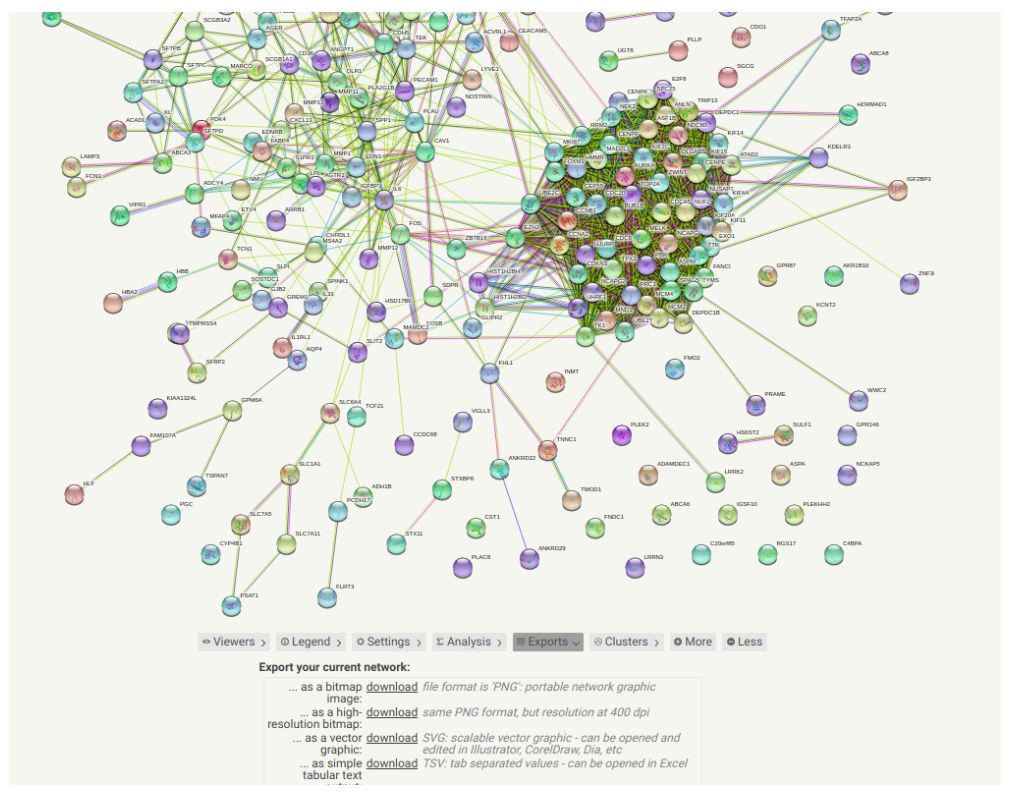
就会导出一个PPI图。有很多圆球和连线。这些又大又圆的球代表的是基因,也可以是蛋白质。在图中,用Node表示。而且那些又细又长的是连接Node的线,叫做edge。edge不仅连接node,而且还有表示interaction的功能。
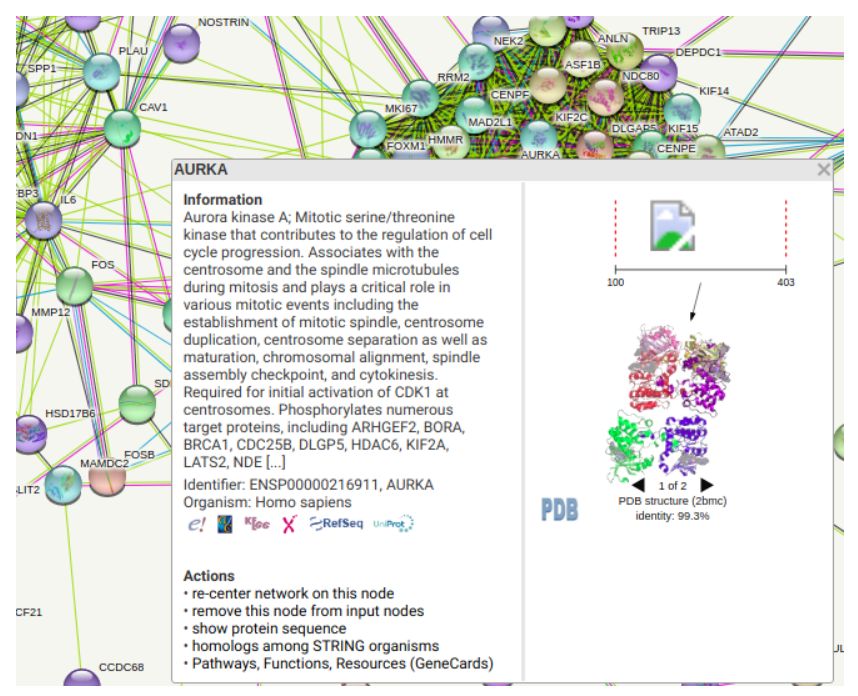
点击Node,还有有相关的信息和域的显示。
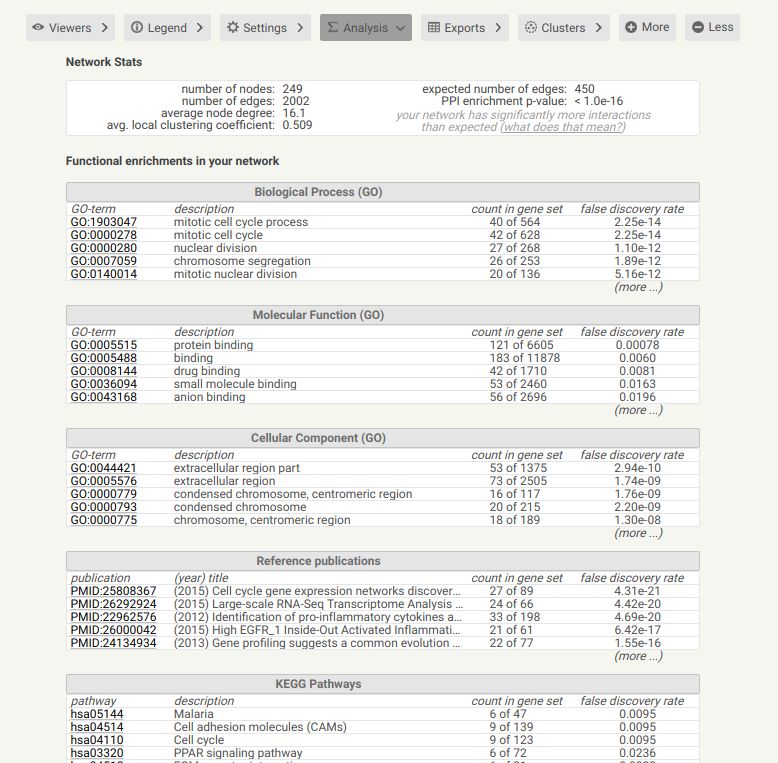
不仅如此,下面的Analysis,还有整个PPI基因/蛋白的GO,KEGG注释。
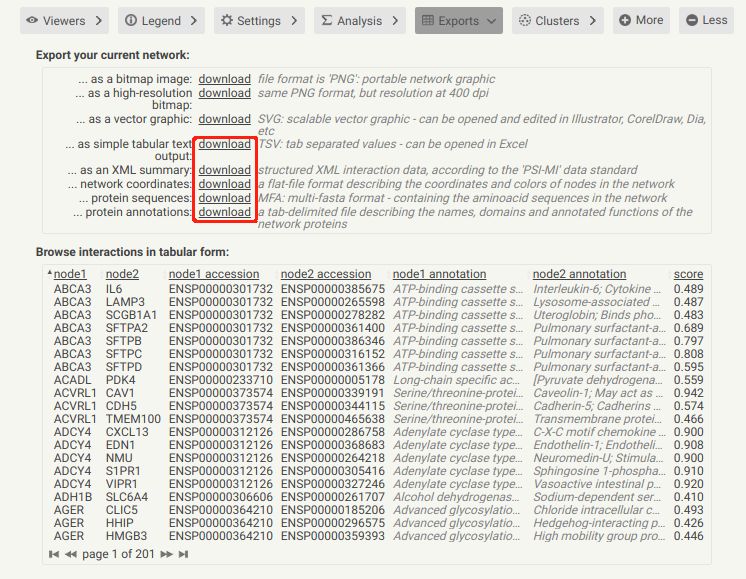
在它输出的文档里面,前面三个download都属于图形文件,下面的三个属于文本文件,可以用来导入cytoscape.可以从下面的表中看到,PPI各个node的关系都已经列好,还对应出每个蛋白ID与注释信息,连它们间的score都有了。这样,就可以基本得到了PPI和比较全面的interaction信息了。
好了,下面的就是从STRING上面下载的5个download文件。可见,下面5个文本文件分别为:string_interaction.tsv(以tab分割);XML 总结;网络坐标;蛋白序列和蛋白注释。应该都可以用excel打开。
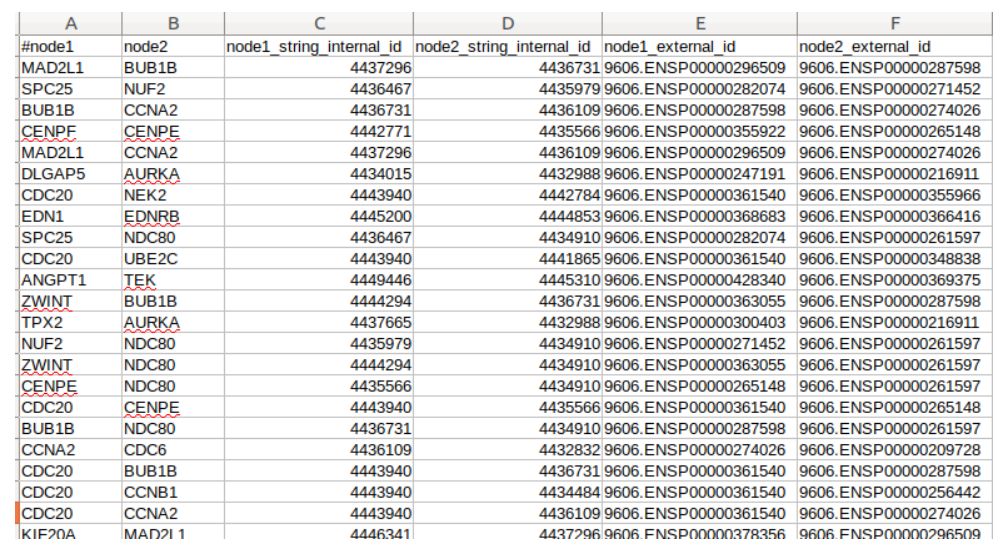
在string_interaction中,有15列,上图为前面6列。第一列为每一个node的基因名,3,5分别是它们对应的内部ID和蛋白ID(这里的蛋白ID还在前面加了物种编号)。2则是和之前那个node有关系的另一个node,4,6也分别是node2的对应内部ID和蛋白ID。
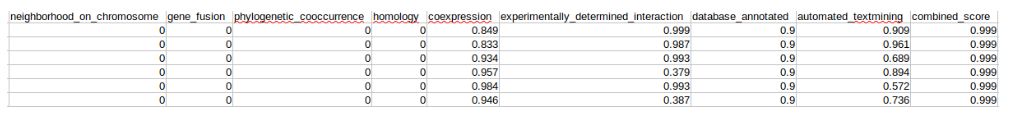
后面9列,分别为染色体上临近点,基因融合,系统发育,同源性,共表达,实验性相互关系,数据库注释,自动文本挖掘,综合评分。
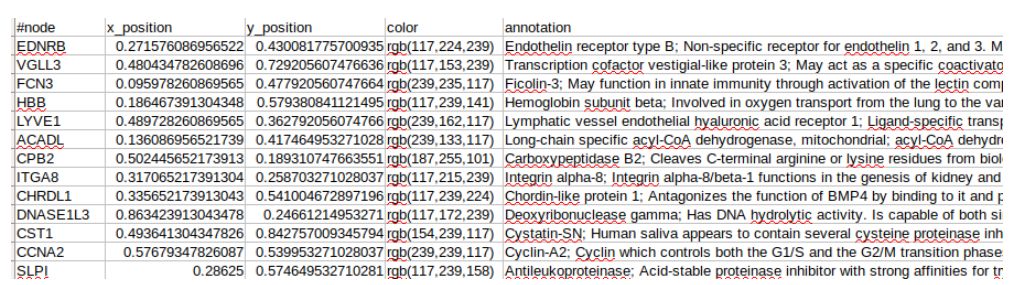
network_coordinate记录的是Node名称,坐标,颜色和注释。

protein_sequences记录的是氨基酸的序列。
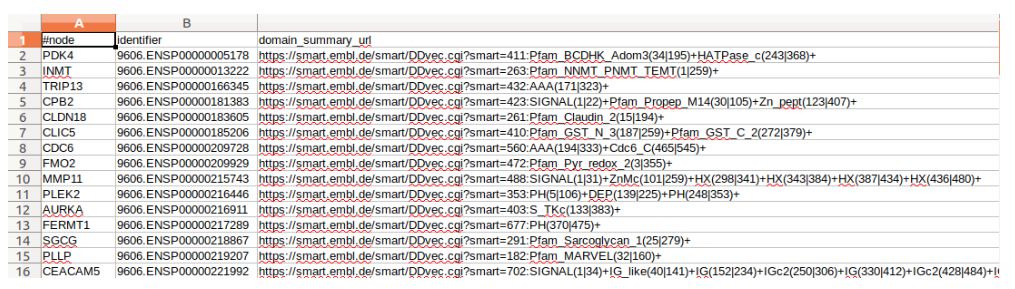
protein_annotations及记录了基因名,蛋白ID和结构域的信息来源。但是由于格式太大,用excel不能完全打开。最后一个XML,是以psimi格式制备的,因此不适宜用excel打开。不然看起来就像cxk打篮球一样。
STRING与R的background gene区别
而在R中,也同样可以对基因进行KEGG/GO注释。那到底哪个更方便,更可信呢。
在R中如何进行注释,这里就不在多说,不知道如何运用R或者还没有试过在R中进行GO/KEGG注释的小伙伴们,可以到JM大神的b站观看视频。
那我们就分别来对比下同样的基因,在STRING和R得到的KEGG/GO注释有啥区别。这里主要是比较STRING和R中的KEGG/GO的background gene库的大小。如果数据库太久或比较小,有很多基因就没有被收录进去,这样有可能我们的目的基因就不会被注释到。(GO注释包括BP,CC和MF)。
基因名如何导入,和网站如何使用,JM大神已经在视频里有详细说明了,而我们就在PPI图下面的exports下载相应的文件就好了。
#注意:在名为‘GeneRation’和‘BgRatio’两列的数据里,我们只看分子。
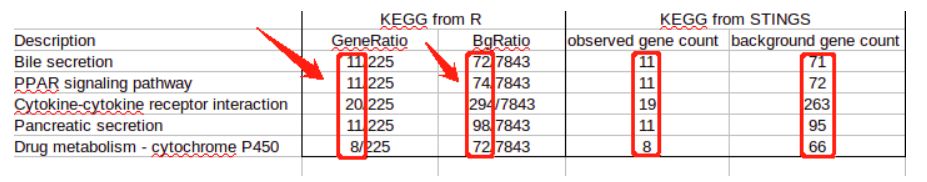
在KEGG注释方面,我们可以看到,各自的区别不大。
那么下面,我们来看看GO中的BP、MF和CC。
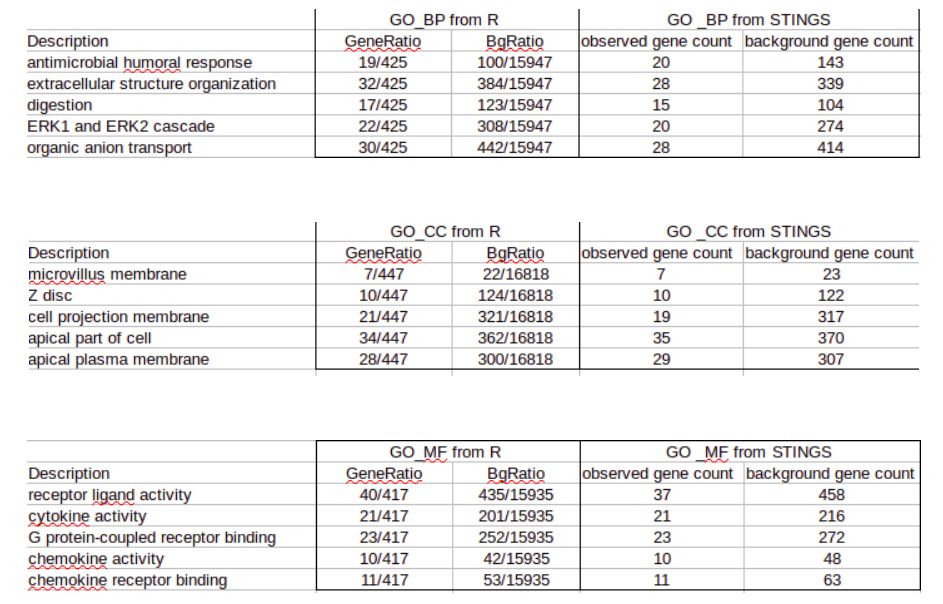
在GO注释方面,同样识别的基因和background区别也不大,所以在KEGG/GO功能注释中,两种方法大家都可以放心使用。
PS:
虽说大多说情况如此,既然可以在STRING这种online tools中做出来的东西,为何我要在R中敲代码来实现呢。
然后,我就发现了某些功能,STRING是很笼统的归为一类,而R中,则会进行比较细致的分类。在这,R中,可以通过p,q值进行cutoff ,而在STRING中,只能通过调节interaction score来cutoff了。所以STRING几秒钟的便捷,和R中细腻还是有一点区别的,看大家所需吧。毕竟鱼与熊掌不可兼得。(但AJ和钢丝球可以)
如果你完全看不懂上面的代码,那么你可能需要下面的课程:
■ ■ ■
(已结束)
(已结束)
(已结束)
(已结束)
最后的最后, 点击下面的阅读原文有惊喜哦!!!
以上是关于GEO数据挖掘流程+STRING VS R in KEGG/GO的主要内容,如果未能解决你的问题,请参考以下文章