R语言GEO数据挖掘-功能富集分析
Posted 医科研
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言GEO数据挖掘-功能富集分析相关的知识,希望对你有一定的参考价值。
欢迎来到医科研,这里是白介素2的读书笔记,跟我一起聊临床与科研的故事, 生物医学数据挖掘,R语言,TCGA、GEO数据挖掘。
 功能富集分析
功能富集分析 
在得到了差异基因的基础之上,进一步进行功能富集分析,这里我们使用clusterprofiler包
本文将对差异基因进行 GO, KEGG注释并完成可视化,GSEA分析
Sys.setlocale('LC_ALL','C')
library(tidyverse)
library(ggplot2)
library(dplyr)
library(clusterProfiler)
library(org.Hs.eg.db)
load(file = "DEG_all.Rdata")
head(DEG)
##定义 筛选logFC>=1, P<=0.05的差异基因
DEG_2<-DEG %>% rownames_to_column("genename") #%>% as_tibble() %>%
filter(abs(logFC)>=0.58,P.Value<=0.05)
genelist<-DEG_2$genename
## ID转换
genelist<- genelist %>% bitr(fromType = "SYMBOL",
toType = c("ENSEMBL", "ENTREZID"),
OrgDb = org.Hs.eg.db)
head(genelist)
dim(genelist)
GO富集分析
注意clusterprofiler中使用的富集ID是ENTREZID,如果不是需要进行转换,也很简单
我们这里其实可以用我们自带的probe中的ID
使用了semi_join这个筛选连接
注意这里我故意使用的是筛选连接,其实inner_join, 等等都可以,我就是要用这些不熟悉的
但是这里的probe其实基因名是有重复的,我们还要进行去重的操作
如果自己有probe信息
if(F){
load("probe.Rdata")
names(probe)[2]<-"genename"
DEG_2<-as_tibble(rownames_to_column(DEG,"genename"))
probe<-probe[!duplicated(probe$genename),]
##设定差异阈值筛选
DEG_2<-DEG_2 %>% arrange(abs(logFC)) %>%
filter(abs(logFC)>=0.58,P.Value<=0.05)
dim(DEG_2)
gene<-probe %>%as_tibble() %>% dplyr::semi_join(DEG_2,by="genename") %>% .$ENTREZ_GENE_ID
head(gene)
length(gene)##相当于是取了交集,用DEG_2中的基因来做筛选,得到了12549个基因
}
我们假设没有probe注释信息
ego <- enrichGO(gene = genelist$ENSEMBL,
OrgDb = org.Hs.eg.db,
keyType = 'ENSEMBL',##指定gene id的类型
ont = "all",##GO分类
pAdjustMethod = "BH",
pvalueCutoff = 0.01,##设置阈值
qvalueCutoff = 0.05,
readable=TRUE)
write.csv(ego,file = "ego.csv")
ego[1:5,1:5]
##
ONTOLOGY ID
GO:0019882 BP GO:0019882
GO:0048002 BP GO:0048002
GO:0019884 BP GO:0019884
GO:0002478 BP GO:0002478
GO:0070482 BP GO:0070482
Description
GO:0019882 antigen processing and presentation
GO:0048002 antigen processing and presentation of peptide antigen
GO:0019884 antigen processing and presentation of exogenous antigen
GO:0002478 antigen processing and presentation of exogenous peptide antigen
GO:0070482 response to oxygen levels
GeneRatio BgRatio
GO:0019882 353/12683 385/19623
GO:0048002 320/12683 347/19623
GO:0019884 314/12683 341/19623
GO:0002478 307/12683 334/19623
GO:0070482 372/12683 420/19623
############
class(ego)
str(ego)
## 三个结果整合在一起
go_dot<-dotplot(ego,split="ONTOLOGY",showCategory=5)+
facet_grid(ONTOLOGY~.)
go_dot
## 单个绘制
ego_MF<-enrichGO(gene = genelist$ENSEMBL,
OrgDb = org.Hs.eg.db,
keyType = 'ENSEMBL',##指定gene id的类型
ont = "MF",##GO分类
pAdjustMethod = "BH",
pvalueCutoff = 0.01,##设置阈值
qvalueCutoff = 0.05,
readable=TRUE)
dotplot(ego_MF,showCategory=6)

KEGG富集分析
代码的风格都是类似的
kk <- enrichKEGG(gene = genelist$ENTREZID,
organism = 'hsa',
pvalueCutoff = 0.05
)
head(kk)
## 打开网页浏览通路
browseKEGG(kk,"hsa04151")
## 点图
dotplot(kk)
## 柱状图
barplot(kk)
点图
dotplot(kk)

柱状图
barplot(kk)

Pathview包用于通路可视化
先调整数据
library(pathview)
pathview(genelist$ENTREZID,pathway.id ="04151",species = "hsa")
##
DEG_2<-DEG %>% rownames_to_column("genename")
DEG_2<-genelist %>% rename(SYMBOL='genename') %>%
inner_join(DEG_2,by='genename') %>%
## select配合everything排序把,改变变量顺序
dplyr::select(ENTREZID,logFC,everything())
head(DEG_2)
###
ENTREZID logFC genename ENSEMBL AveExpr t P.Value
1 218 -3.227263 ALDH3A1 ENSG00000108602 10.302323 -10.710306 4.482850e-05
2 1545 -3.033684 CYP1B1 ENSG00000138061 13.287607 -10.505888 4.995753e-05
3 1543 -9.003353 CYP1A1 ENSG00000140465 11.481268 -8.371476 1.762905e-04
4 11148 -1.550587 HHLA2 ENSG00000114455 6.595658 -7.443431 3.337672e-04
5 8140 -2.470333 SLC7A5 ENSG00000103257 13.628775 -7.298868 3.708688e-04
6 25976 -1.581274 TIPARP ENSG00000163659 12.764218 -7.024252 4.552834e-04
adj.P.Val B
1 0.3134585 -4.048355
2 0.3134585 -4.049713
3 0.7374232 -4.069681
4 0.9308066 -4.083411
5 0.9308066 -4.085966
6 0.9522252 -4.091192
###
构建geneList对象
这是比较关键的一步,需要构建geneList对象,至少需要包括ID与排序好的数值型变量
数值变量降序排列
sign.pos参数包括的位置有4种:"bottomleft", "bottomright", "topleft" and "topright".
geneList<-DEG_2$logFC
names(geneList)<-as.character(DEG_2$ENTREZID)
geneList<-sort(geneList,decreasing=TRUE)
#geneList<-geneList[!duplicated(names(geneList))]
head(geneList)##构建geneList对象成功
1580 7025 54474 9247 56603 4907 ## geneid
2.378155 2.365342 2.258682 2.180382 2.143749 2.030780 ## logFC
## 这样logFC值就能反应在通路的变化中了
pathview(geneList,pathway.id ="04151",species = "hsa",same.layer = F )

调整函数的参数,改变图形
比较重要的是kegg.native = F,可以调整输出为矢量图
## 看不出变化的参数kegg.native,默认设置为T
pathview(geneList,pathway.id ="04151",species = "hsa",
out.suffix = "KEGG",##输出文件后缀
kegg.native = T,
same.layer = F )
## 可以产生矢量图pdf文档,
pathview(geneList,pathway.id ="04151",species = "hsa",
out.suffix = "KEGG",##输出文件后缀
kegg.native = F,
split.group = T,#是否分开节点组
sign.pos= "topleft",##图注的位置
same.layer = F )

GSEA分析
GSEA分析在高分文章中还是一个很常见的分析,之前认为官方软件的作图达不到很高的要求,现在终于可以解决了clusterprofiler包更新的画图功能模仿官网的图,颜值更高,操作更简单,大家都值得拥有。
KEGG的GSEA富集分析
head(geneList)
##
1580 7025 54474 9247 56603 4907
2.378155 2.365342 2.258682 2.180382 2.143749 2.030780
##
#geneList<-geneList[!duplicated(geneList)]
kk2<- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 1000,
minGSSize = 120,
pvalueCutoff = 0.5,
verbose = FALSE)
head(kk2)
##
ID Description setSize enrichmentScore
hsa04080 hsa04080 Neuroactive ligand-receptor interaction 293 -0.3593278
hsa04024 hsa04024 cAMP signaling pathway 185 -0.3656357
hsa04060 hsa04060 Cytokine-cytokine receptor interaction 244 -0.3358228
hsa04020 hsa04020 Calcium signaling pathway 172 -0.3566872
hsa04062 hsa04062 Chemokine signaling pathway 165 0.3112935
hsa05202 hsa05202 Transcriptional misregulation in cancer 163 -0.3537420
NES pvalue p.adjust qvalues rank
hsa04080 -1.481227 0.002649007 0.1562914 0.1422098 1912
hsa04024 -1.426245 0.007062147 0.2083333 0.1895629 1697
hsa04060 -1.356610 0.012345679 0.2427984 0.2209227 2055
hsa04020 -1.378406 0.017118402 0.2524964 0.2297470 1748
hsa04062 1.323383 0.025806452 0.2676695 0.2435530 1332
hsa05202 -1.361718 0.027220630 0.2676695 0.2435530 2463
leading_edge
hsa04080 tags=31%, list=16%, signal=27%
hsa04024 tags=26%, list=14%, signal=23%
hsa04060 tags=30%, list=17%, signal=25%
hsa04020 tags=26%, list=15%, signal=22%
hsa04062 tags=19%, list=11%, signal=17%
hsa05202 tags=32%, list=21%, signal=26%
##
GSEA图可视化
之前的图确实颜值不够
gseaplot(kk2,geneSetID = "hsa04080")

新功能gseaplot2-模仿GSEA软件的图-确实更好看了
library(enrichplot)
gseaplot2(kk2,##KEGG gse的对象
geneSetID = "hsa04080",
pvalue_table=T)

修改线条颜色
现在的图-高端大气上档次
gseaplot2(kk2,##KEGG gse的对象
geneSetID = "hsa04080",
color="red",
pvalue_table=T)
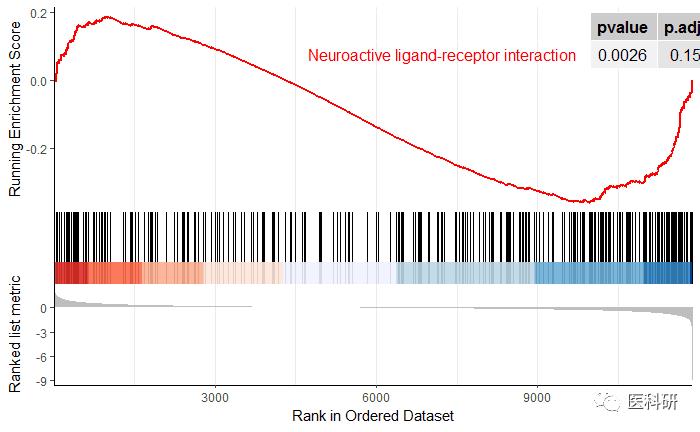
MgsiDB的GSEA富集分析
gmtfile <- system.file("extdata", "c5.cc.v5.0.entrez.gmt", package="clusterProfiler")
c5 <- read.gmt(gmtfile)
gene <- names(geneList)
egmt <- enricher(gene, TERM2GENE=c5)
head(egmt)
##
ID Description GeneRatio BgRatio
CELL_FRACTION CELL_FRACTION CELL_FRACTION 451/4461 493/5270
MEMBRANE_FRACTION MEMBRANE_FRACTION MEMBRANE_FRACTION 309/4461 339/5270
pvalue p.adjust qvalue
CELL_FRACTION 1.764189e-06 0.0003775364 0.00036398
MEMBRANE_FRACTION 1.870439e-04 0.0200136995 0.01929506
##
#write.csv(egmt,file = "egmt.csv")
GSEA
egmt2 <- GSEA(geneList, TERM2GENE=c5,
pvalueCutoff = 0.5,#
verbose=FALSE)
head(egmt2)
gseaplot2(egmt2, geneSetID = 1, title = egmt2$Description[1],pvalue_table=T)

还可以同时画多个
gseaplot2(egmt2, geneSetID = 1:3,pvalue_table=T)

Gene-Concept Network
这里的输入egmt2可以是ego,kk,edo都可以,进行可视化
edox <- setReadable(egmt2, 'org.Hs.eg.db', 'ENTREZID')
cnetplot(edox, foldChange=geneList)

换个输出形状
cnetplot(edox, foldChange=geneList, circular = TRUE, colorEdge = TRUE)

One more thing
• 如果你对以上这些内容完全看不懂,想了解数据库挖掘,欢迎关注我录制的视频课程 零代码数据挖掘与案例解析,几十个精华在线数据库一网打尽。下方可以扫码直达或者点击阅读原文可直达课程:


组建科研交流群通知
优秀会吸引优秀,微光会吸引微光。

欢迎志同道合的朋友加群交流,白介素2同学邀请您一起学习讨论(注:添加本人微信请先备注姓名单位等基本信息,否则不会通过)。
相关阅读
本期内容就到这里,我是白介素2,下期再见,点击下方框框留言。
交易担保 推文留言 点击留言 Mini Program
更多精彩扫一扫
你来或不来,我就在那里
编辑:林芝
以上是关于R语言GEO数据挖掘-功能富集分析的主要内容,如果未能解决你的问题,请参考以下文章