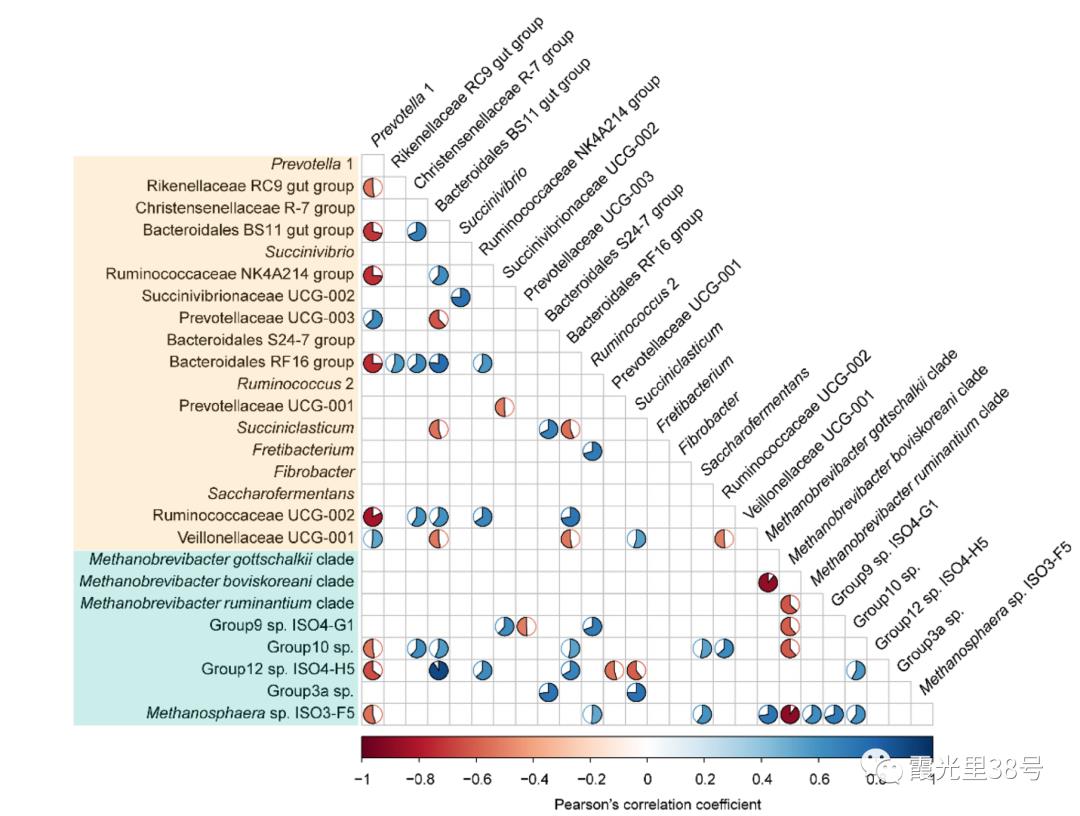
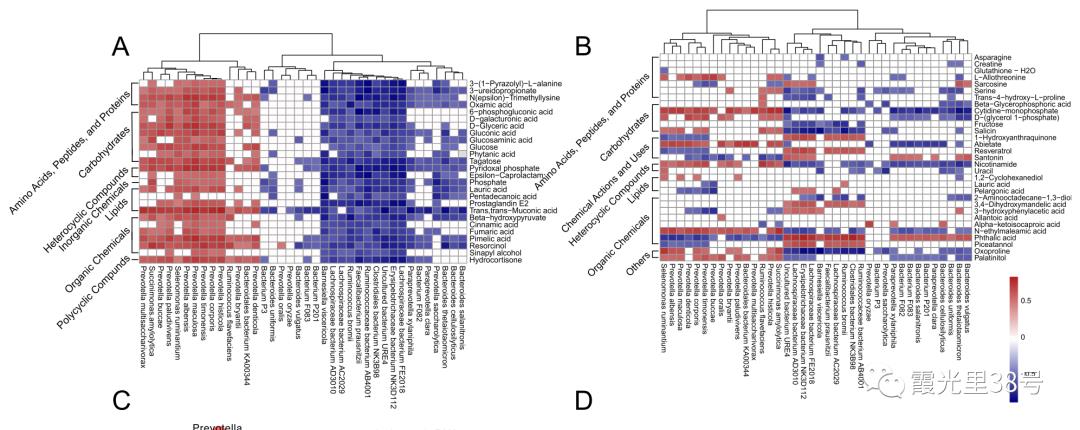
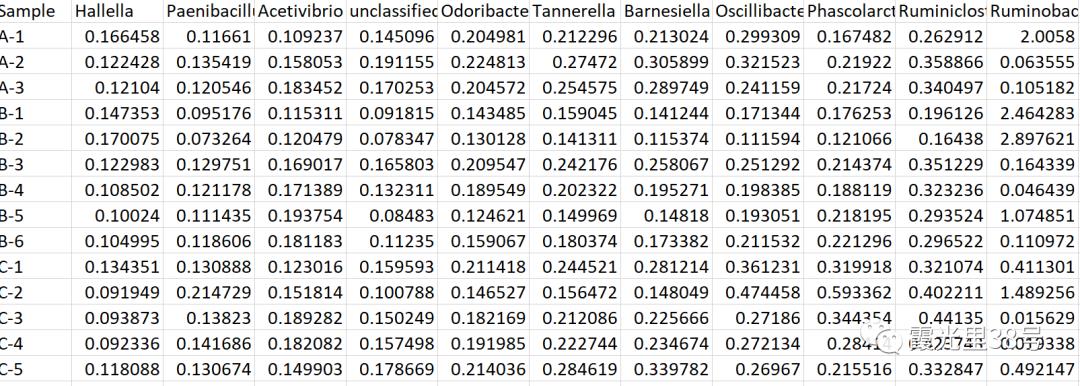
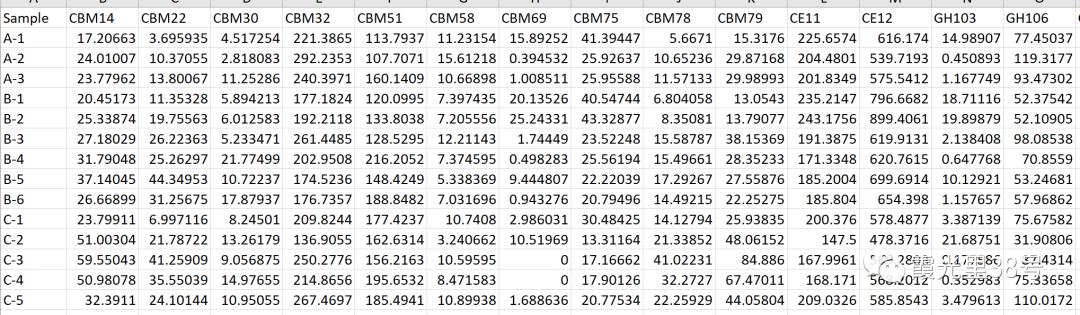
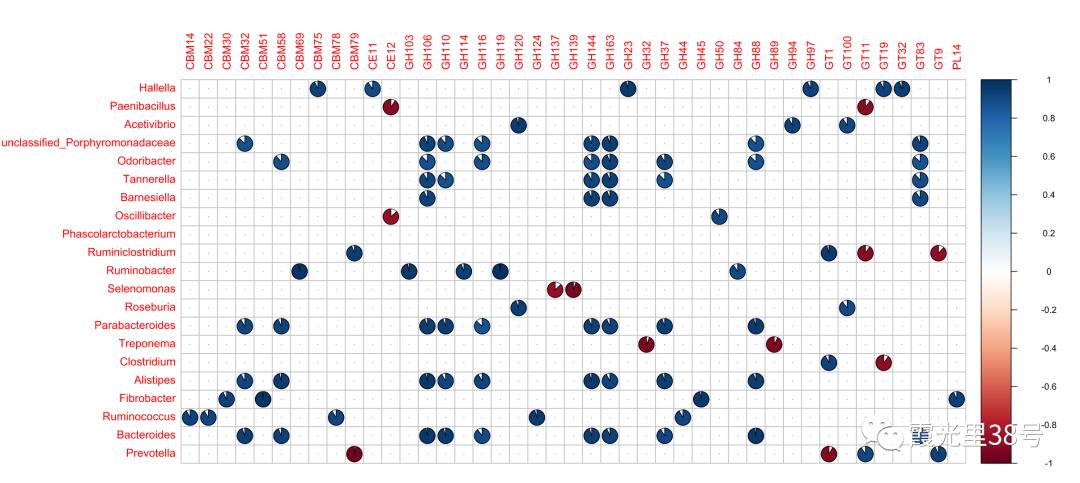
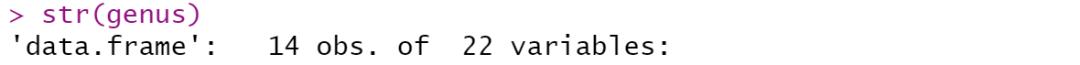相关分析就是对总体中确实具有联系的标志进行分析,其主体是对总体中具有因果关系标志的分析。它是描述客观事物相互间关系的密切程度并用适当的统计指标表示出来的过程。在一段时期内出生率随经济水平上升而上升,这说明两指标间是正相关关系;而在另一时期,随着经济水平进一步发展,出现出生率下降的现象,两指标间就是负相关关系。 为了确定相关变量之间的关系,首先应该收集一些数据,这些数据应该是成对的。例如,每人的身高和体重。然后在直角坐标系上描述这些点,这一组点集称为“散点图”。根据散点图,当自变量取某一值时,因变量对应为一概率分布,如果对于所有的自变量取值的概率分布都相同,则说明因变量和自变量是没有相关关系的。反之,如果,自变量的取值不同,因变量的分布也不同,则说明两者是存在相关关系的。两个变量之间的相关程度通过相关系数r来表示。相关系数r的值在-1和1之间,但可以是此范围内的任何值。正相关时,r值在0和1之间,散点图是斜向上的,这时一个变量增加,另一个变量也增加;负相关时,r值在-1和0之间,散点图是斜向下的,此时一个变量增加,另一个变量将减少。r的绝对值越接近1,两变量的关联程度越强,r的绝对值越接近0,两变量的关联程度越弱。 一般我们比较常见的是组内微生物与微生物之间的相关性分析如在文章Methane Emission, Rumen Fermentation, and Microbial Community Response to a Nitrooxy Compound in Low-Quality Forage Fed Hu Sheep,作者在属水平上进行微生物之间的相关性分析。但是在实际情况中,尤其是多组学关联分析的时候,往往是两个数据集之间的相关性分析。如2020年在线发表在Microbiome(IF>11)的一篇文章Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance,作者进行了瘤胃宏基因组-菌群丰度与血清、瘤胃内容物的代谢组关联分析:接下来,我们在R中运行相关性分析,计算微生物-环境因子间的相关性。我们需要把微生物相对丰度和环境因子(本文为CAZymes的基因丰度CPM),整合在一块儿。属水平的微生物相对丰度,genus