R语言mRNA差异表达分析
Posted 易学统计
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言mRNA差异表达分析相关的知识,希望对你有一定的参考价值。

上一篇文章介绍的是mRNA数据的整理,整理完就开始介绍的是如何用R语言来做差异表达分析,继续码字。
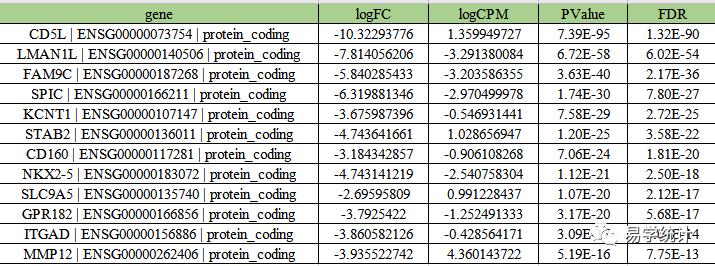
以上这个表格,在表达差异分析中很常见,第一列是基因名称。
第二列是基因的logFC值,FC是即fold change,表示实验组比上对照组的差异表达倍数。国内的一般默认取其对数绝对值大于2作为差异基因的筛选标准,这个阈值是可以根据实际情况进行调控。
第三列是logCPM,CPM是 Counts Per Million,是每个基因一个指标。
第四列是P值,其零假设是这个基因的表达在正常组织和癌症组织上是一样的,因此P<0.05可认为某个基因在两组间表达有显著差异。
第五列FDR是对P值的校正结果,一般使用FDR小于0.05。
火山图数据使用:FDR取对数和FC取对数,用来绘制火山图,并标注上调和下调基因。其中上调是指FC取对数大于某个正数,比如1,下调指的是FC取对数小于某个负数,比如-1.
热图数据使用:根据每个样本中每个基因的表达量绘制热图。其中每个小方格表示每个基因,颜色表示该基因表达量的大小。表达量越大,颜色越深(一般红色为上调,绿色为下调)
根据样本名称区分正常组织和癌症组织。首先加载上次处理后的数据集,上次用两种方法生成两个数据集,本次我们以第一个数据集为例子。
根据第一篇文章介绍01-09表示肿瘤样本,10-19表示正常对照组织。将癌症样本归为'tumor',正常对照组织归为'normal'。
setwd('~\下载数据\')load(file = "mRNA_exprSet.Rda")###metadata <- data.frame(names(mRNA_exprSet)[-1])for (i in 1:length(metadata[,1])) {num <- as.numeric(substring(metadata[i,1],14,15))if (num %in% seq(1,9)) {metadata[i,2] <- "tumor"}if (num %in% seq(10,29)) {metadata[i,2] <- "normal"}}
打开基因数据这个文件夹 可以看到里面有182个文件夹,每一个文件夹里面还有1个压缩文件。这里面其实就是下载的182个样本,每个样本的信息单独保存在一个文件夹。这种数据不能直接拿来做分析,因此先要合并。
mycounts <- mRNA_exprSetmycounts=as.matrix(mycounts) ##DGEList对象要求矩阵格式rownames(mycounts)=mycounts[,1]exp=mycounts[,2:ncol(mycounts)]dimnames=list(rownames(exp),colnames(exp))data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)data=avereps(data) ##如果有重名的基因,就自动取平均值,方便后面的分析data=data[rowMeans(data)>1,] ##这个基因在所有的样品上基本都不表达或者是低表达,就删除。
预处理前是19668个基因,删除在所有样品上基本不表达或者低表达的基因,剩下17922个基因。
也可以采用cpm校准化的基因表达量作为过滤低表达基因的标准。
group <- as.vector(metadata$sample) #按照癌症和正常样品数目修改design <- model.matrix(~group) ##变成一个实验设计对象y <- DGEList(counts=data,group=group) ##变成edgr包可以识别的数据结构,data需要是矩阵格式
edgeR这个包将数据存储在DGEList对象中,需要指定的参数包括counts 和 group。其中表达矩阵存储在$counts中,样本分组信息等存储在$samples中。
如果采用cpm过滤基因,R代码操作如下:
# cpm normalizationcountsPerMillion <- cpm(y)countCheck <- countsPerMillion > 1keep <- which(rowSums(countCheck) >= 2)<- y[keep,]##16889 182
①构建完DGEList对象,做因子校正
y <- calcNormFactors(y) ##对因子进行校正edgeR默认使用TMM(trimmed mean of M-values)进行normalization,以最大程度地缩小样本间基因表达量的log-fold change。这是因为TMM 法认为样本间大部分的基因都没有发生差异表达,而那些真正差异表达的基因并不会受到normalization的严重影响。如此一来,便将那些由于测序引起的差异表达基因的表达量给校正了,消除了一部分的假阳性。
为什么要做Normalization呢?
在差异分析中,我们常常更关注的是相对表达量的变化,例如处理组的A基因表达量相对于对照组的而言是上调还是下调了。而基因表达量的定量准确性则在差异分析中不太重要,因此,在进行差异分析时,像RPKM/FPKM这种对转录本长度进行normalization方法是并不常用,也是没有必要的。
在常规的RNA-seq中,影响基因表达量更大的技术因素往往是测序深度以及有效文库大小(effective libraries size)。这也是一般的差异分析软件会进行normalize的部分
y <- estimateCommonDisp(y) ##为所有基因都计算同样的dispersiony <- estimateTrendedDisp(y) ##根据不同基因的均值--方差之间的关系来计算dispersion,并拟合一个trended modely <- estimateTagwiseDisp(y) ##计算每个基因的dispersion,并利用经验性贝叶斯方法(empirical bayes)压缩至Trend Didspersion。##个人认为这一项相当于GLM中每个基因的beta值plotBCV(y)
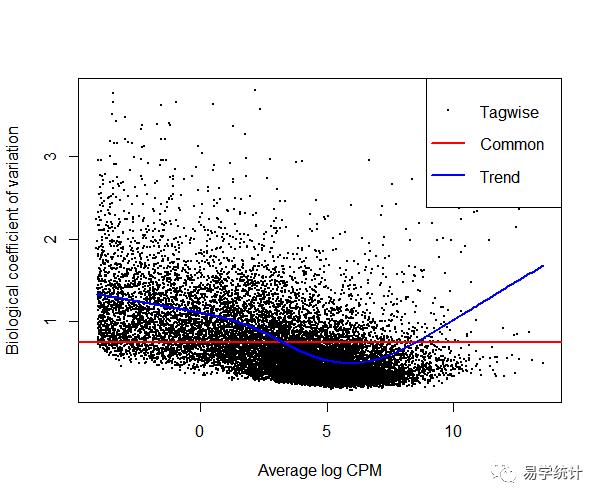
③离散值估计的结果解释:
关于dispersion estimation
一般而言,样本间的变异系数(coefficient of variance,CV)是由两部分组成的,一是技术差异(Technical CV),另一个是生物学差异(Biological coefficient of variance,BCV)。前者是会随着测序通量的提升而消失的,而后者则是样本间真实存在的差异。所以,若一个基因的BCV值在样本间比较大的话,可认为这是个差异表达基因。而edgeR正是通过估计dispersion来估计BCV,进而拟合出线性回归模型的参数。
BCV是负二项分布的平方根。 该图显示BCV估计值和趋势估计值。
这里引出了负二项分布,为什么用这个分布?
在基因组学中,基因表达的reads数,可以看作一个计数的过程,我们可以把采样自某个基因的reads数量建模为泊松分布,并利用泊松回归估计具体的数量。
然而,对于同一个基因来说,通常var比mean要大(比如进行了3次生物学重复实验,假设测了3只同样疾病状态的小鼠;注意,生物重复不是技术重复;这3次得到某基因的reads counts,一般来讲方差大于其均值),特别是对于高表达的基因。因此,负二项回归要更合适一点。
Poisson分布与负二项分布有什么关系呢?Poisson分布中,设定μ是常数,当μ不是常数,而是一个随机变量,且服从γ分布时,此时复合Poisson分布就是负二项分布。
而负二项分布(NB)需要均值和离散值两个参数。edgeR对每个基因都估测一个经验贝叶斯稳健离散值(mpirical Bayes moderated dispersion),还有一个公共离散值(common dispersion,所有基因的经验贝叶斯稳健离散值的均值)以及一个趋势离散值,见上代码注释。

④:显著性差异检验:
et <- exactTest(y,pair = c("normal","tumor")) #检验两组时间的变异系数## ##类似配对T检验ordered_tags <- topTags(et, n=100000)##P值的校正 采用BH方法
这个执行完就是出来文章开头讲的表格,差异表达的工作已经完成,后面的工作就是基于这个表格做一些基因的筛选和图片的绘制。
allDiff=ordered_tags$tableallDiff=allDiff[is.na(allDiff$FDR)==FALSE,]diff=allDiff ##提取所有基因差异分析后的结果,见下表1newData=y$pseudo.counts ##提取校准后表达的数据,见下表2
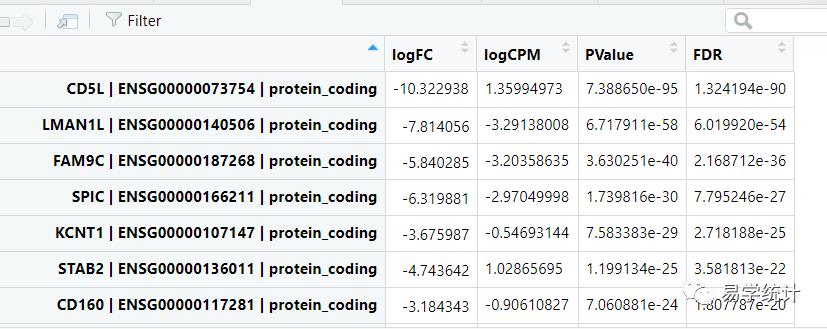
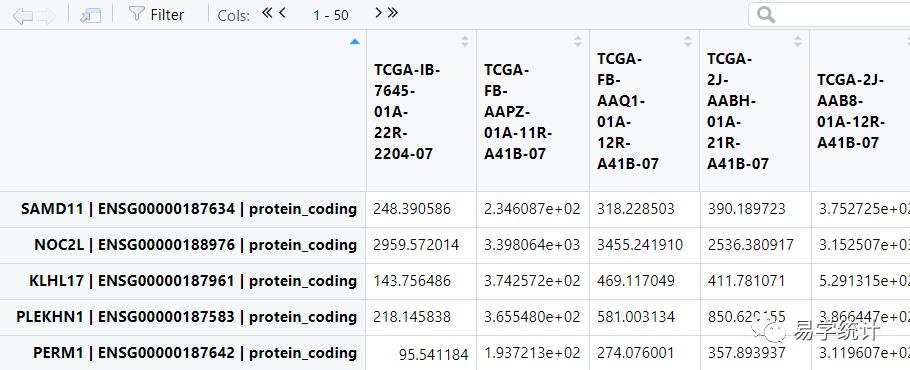
②筛选
###定义两个筛选条件foldChange=1 #差异在2倍以上padj=0.05 ##校正后的P值小于0.05###筛选P值小于0.05且logFC的绝对值大于1的基因diffSig = diff[(diff$FDR < padj & (diff$logFC>foldChange | diff$logFC<(-foldChange))),]write.table(diffSig, file="diffSig.xls",sep=" ",quote=F)## 662 4###显示上调基因###筛选P值小于0.05且logFC的大于1的基因diffUp = diff[(diff$FDR < padj & (diff$logFC>foldChange)),]write.table(diffUp, file="up.xls",sep=" ",quote=F)## 36 4##显示下调基因###筛选P值小于0.05且logFC的小于-1的基因diffDown = diff[(diff$FDR < padj & (diff$logFC<(-foldChange))),]write.table(diffDown, file="down.xls",sep=" ",quote=F)## 626 4###输出所有基因校正后的表达normalizeExp=rbind(id=colnames(newData),newData)write.table(normalizeExp,file="normalizeExp.txt",sep=" ",quote=F,col.names=F)#输出差异基因校正后的表达diffExp=rbind(id=colnames(newData),newData[rownames(diffSig),])write.table(diffExp,file="diffmRNAExp.txt",sep=" ",quote=F,col.names=F)heatmapData <- newData[rownames(diffSig),]
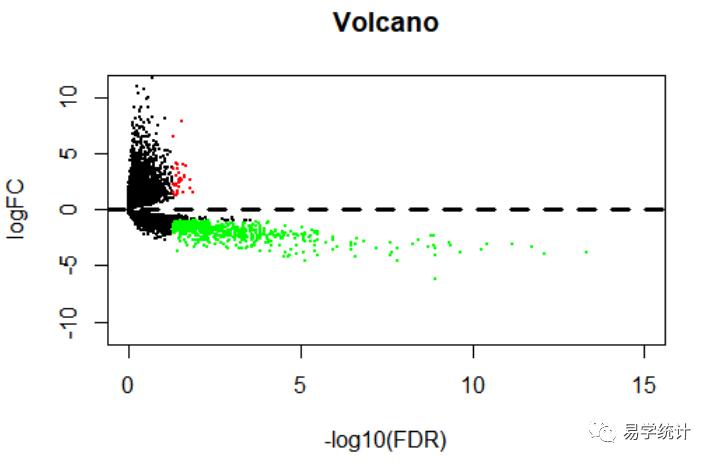
火山图用途?
下图1中,火山图可反映总体基因的表达情况,横坐标代表log2(Fold Change),纵坐标表示-log10(P值),值越大表示差异越显著,每个点代表一个基因,绿色点是下调2倍以上的基因,红色点是上调2倍以上的基因。处于二者之间的标记为黑色。
下面我们看一下火山图的绘制。
pdf(file="vol.pdf")xMax=max(-log10(allDiff$FDR))+1yMax=12plot(-log10(allDiff$FDR), allDiff$logFC, xlab="-log10(FDR)",ylab="logFC",main="Volcano", xlim=c(0,xMax),ylim=c(-yMax,yMax),yaxs="i",pch=20, cex=0.4)diffSub=allDiff[allDiff$FDR<padj & allDiff$logFC>foldChange,]points(-log10(diffSub$FDR), diffSub$logFC, pch=20, col="red",cex=0.4)diffSub=allDiff[allDiff$FDR<padj & allDiff$logFC<(-foldChange),]points(-log10(diffSub$FDR), diffSub$logFC, pch=20, col="green",cex=0.4)abline(h=0,lty=2,lwd=3)dev.off()
聚类热图用途?
横坐标代表样本聚类,一列代表一个样本,聚类基于样本间基因表达的相似性,样本间基因表达越接近,靠的越近,以此类推。
纵坐标代表基因聚类,一行代表一个基因,聚类基于基因在样本中表达的相似性,基因在样本中表达越接近,靠的越近,以此类推。
色阶代表基因表达丰度,越红代表上调得越明显,越绿代表下调得越明显。这图一般做展示数据用,看上去直观点。
hmExp=log10(heatmapData+0.001)library('gplots')hmMat=as.matrix(hmExp)pdf(file="heatmap.pdf",width=60,height=90)par(oma=c(10,3,3,7))heatmap.2(hmMat,col='greenred',trace="none")dev.off()
本次完成基因差异的分析和两个常见图的绘制,下期分享共表达。
作者介绍:医疗大数据统计分析师,擅长R语言。
欢迎各位在后台留言,恳请斧正!
更多阅读
长按二维码
易学统计
以上是关于R语言mRNA差异表达分析的主要内容,如果未能解决你的问题,请参考以下文章