序列比对-动态规划算法
Posted 生信之旅
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了序列比对-动态规划算法相关的知识,希望对你有一定的参考价值。
在研究生物学功能的时候,一般同源性序列意味着相似性,虽然序列相似不一定可以说明序列同源,但是相似度高的序列很有可能也是同源的,这一假设对研究基因和蛋白质很有帮助。一对残基间可能的比对只有三种,
序列X的碱基A比对到序列Y的碱基B;
序列X的碱基A比对到序列Y的空位;
序列X的空位比对到序列Y的碱基B;
下面基于此介绍一下比对的方法。
序列X: AAG
序列Y: AGC
一、点阵图
使用点阵图的形式来表示序列比对是比较直观的,下面我们使用序列ATAT与其本身进行比对来进行说明,其中行与列元素相同的为1,不同则为0,得到如下矩阵。
从上面的矩阵我们可以发现:
对角线为1表明全匹配上了,即ATAT;
平行于对角线的,是子序列匹配上了,即右上角的AT;
平行于反对角线的,说明反向互补,即ATA;
二、全局比对(needleman-wunsch算法)
若我们想要得到两条序列间最佳比对结果的话,我们一般想到的都是穷举法。但是此方法需要穷举的次数随着序列长度的增加而不断增大。假设我们需要比对的两条序列均为n,则比对结果的长度范围为n(两条序列完全匹配上)-2n(两条序列均对到空位),从上述可以推导,两条长度为n的序列之间产生的任何一个比对结果,都可以在2n个可能中放置n个空位产生,即: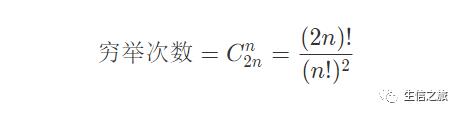
从上述式子可以看出,比对序列的长度越长,需要穷举的次数越多,这对我们来说将会耗时很多,这是不太现实的。所以我们需要一种新的方法进行比对——动态规划。动态规划算法是一种求取最优解的方法,该方法通过将问题拆解为若干子问题,求取子问题的最优解,所有子问题的最优解合并即为全局最优解。
下面将以序列X、Y为例,简单讲解该算法原理。
首先,我们做出如下假设:

则比对算法应为: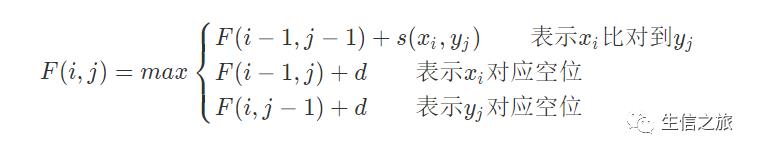
对于以上三个公式的理解是(0≤i≤n,0≤j≤n,n为序列长度):

为了方便说明,这里我们引入一个打分矩阵,同时定义gap open(d) = gap extend(e) = -5,同时以矩阵的形式显示比对过程。
打分矩阵一般基于考虑碱基(氨基酸)理化性质等因素设定的,常用的有(DNA打分矩阵:等价矩阵、转换-颠换矩阵、Blast矩阵;蛋白质打分矩阵:PAM、Blosum)
本例使用的打分矩阵如下: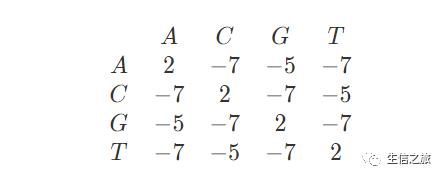
根据上面的公式及打分矩阵,我们可以得到以下的数组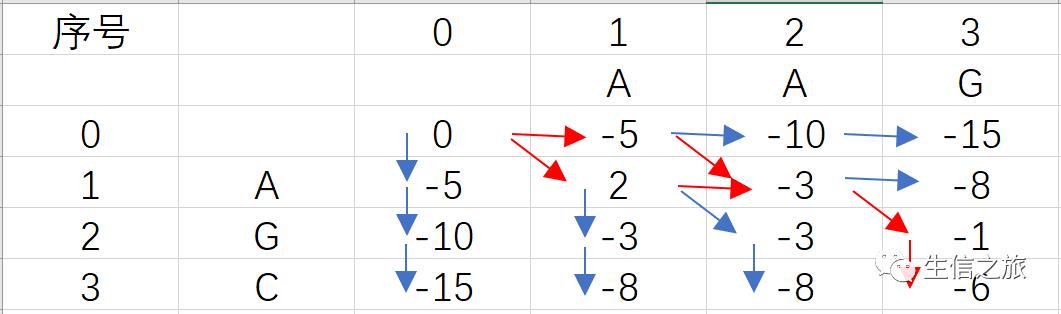
其中箭头表示分数的来源,红色箭头指示的路径为最优。下面简单说明下各个分数的计算过程。

回溯过程也很简单,从矩阵的右下角开始往上回溯,即红色箭头部分,其中
从斜对角线来源的箭头表示碱基匹配;
从上面来源的箭头表示序列Y比对到空位;
从左边来源的箭头表示序列X比对到空位;
最终我们得到的比对结果为
AAG-
-AGC
与
AAG-
A-GC
这两个结果都是最优比对结果,分数均为-6
三、局部比对(smith-waterman算法)
上面讲的是全局比对,即查询序列与目标序列都是从头到尾的全部碱基进行比对。该算法在早期的蛋白序列比对中应用广泛,但是随着生物序列越来越多,越来越长,这种比对算法遇到了问题,即研究人员发现功能相关的蛋白之间虽然整体的序列相差甚远,但是常常具有相同的功能域。对于这种情况,仅靠全局比对算法是不行的;另一方面,70年代内含子的发现,使得在进行核酸序列比对时,必须要能正确处理内含子导致的大片段差异。所以我们需要新的方法来发现局部相似序列。
局部比对算法设计很巧妙,仅在全局比对算法的基础上增加了一个条件,即限制了最低分数为0,公式如下所示:
我们这里同样使用上诉的例子来进行说明,得到如下的矩阵:
从上图可以看出,与全局比对相比,有以下几点不同:
最小值为0,没有负数了;
存在多条独立的路径;
回溯的起点不是右下角,终点也不一定是左上角;
其实,引入一个‘止损’下限分数,其实质是提供了在差异较大区域重启比对的能力,从而可以有效的发现局部的相似性。
以上是关于序列比对-动态规划算法的主要内容,如果未能解决你的问题,请参考以下文章