文献速递:神经网络算法&计算化学中从头算镓的成核与相图 Posted 2021-04-24 模拟计算联盟
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了文献速递:神经网络算法&计算化学中从头算镓的成核与相图相关的知识,希望对你有一定的参考价值。
作者介绍:
Michele Parrinello
米歇尔-帕里内洛(Michele Parrinello)是非常出名的分子动力学研究的意大利物理学家。帕里内罗(Parrinello)和罗伯托·卡尔(Roberto Car)凭借对1985年开创性论文“分子动力学和密度泛函理论的统一方法”首次提出的Car-Parrinello方法的不断发展,于2009年荣获狄拉克奖章和西德尼·芬巴赫奖。
标题:Ab initio phase diagram and nucleation of gallium
1. 元素镓具有一些有趣的特性,如低熔点、密度异常和共价与金属共存的电子结构。为了模拟这个复杂的系统,研究者通过对多热-多压模拟中产生的构型进行密度泛函理论计算,训练神经网络来构造了从头算质量相互作用势。
2. 在这里,研究者分析表明,液体镓,α-镓,β-镓,镓-II之间的相对平衡是很好的描述,所得相图与实验结果一致。
3. 此外,研究者还描述了液态镓的局域结构及其在α-镓和β-镓中的成核,研究表明,亚稳态β-Ga的生成比热稳态α-Ga的生成更有利。
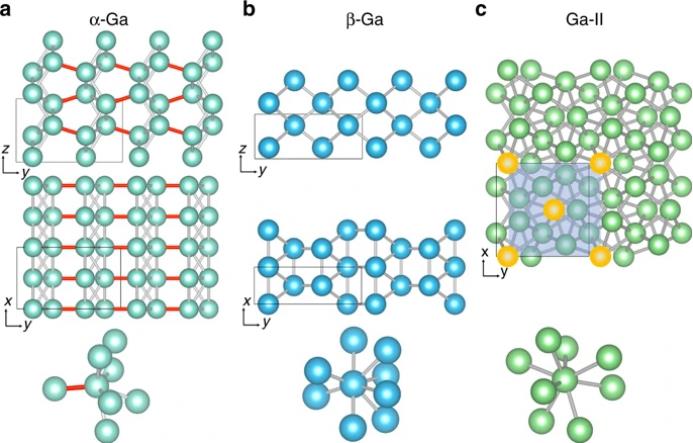
元素镓是一种独特的金属,具有许多迷人和不寻常的特性。在它的不同相中,发现了其具有各种重要的技术应用。不像大多数金属,包括那些在元素周期表同一列的金属,它在相当复杂的结构中结晶,如图1所示。它在环境条件下的稳定固相,称为α-Ga,是正交的,在原始单胞中有四个原子。每个原子与七个相邻原子相互协调,形成一个各向异性极强的原子环境(图1a)。最邻近的两个原子之间的成键被称为共价键,因而α-Ga被认为是共价键和金属键的混合物。
在融化时,像水一样,它表现出密度异常,液体膨胀高达3.1%。尽管经过了几十年的实验研究,
液体镓的结构仍未被完全理解
。此外,微米级或亚微米级的液态镓可以过冷至150k而不凝固。在这种情况下,晶化过程中不会产生稳定的晶相,而会产生大量的β-Ga结构。实验还表明,在3-15 nm范围内的Ga纳米颗粒可以过冷,甚至可以降至90 K,没有冻结转变的迹象。β-Ga的结构是单斜的,包含了平行于(021)平面的原子的正方形排列,如图1b所示。与α-Ga不同,在距离为2.78 Å的情况下,β-Ga中的每个原子有8个最近邻。此外,Ga相图显示了各种不同的复杂稳定和亚稳态多晶型,例如,δ-Ga,γ-Ga和Ga-II相。其中Ga-II在高压下具有热力学稳定性,可以描述为每个原子具有8倍配位的互穿体中心立方晶格(图1c)。
液体和固体Ga结构
的
复杂性
使得
分子模拟的成核
和
相对平衡的研究
具有
很大的挑战性
。
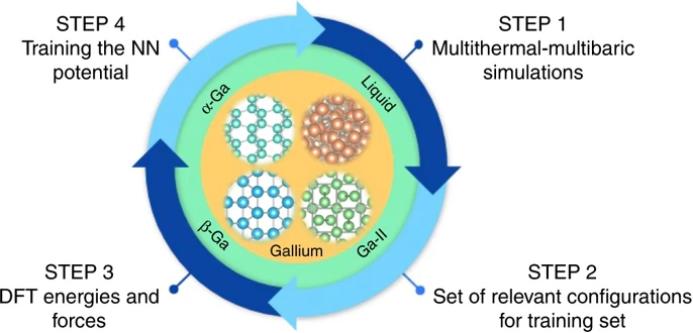
为了进行模拟,需要清除两个障碍。一个是 成核发生的长时尺度,而另一个是对导致遗传算法复杂行为的微妙相互作用的精确描述。幸运的是,这两个领域都取得了很大进展。一方面,高效的强化取样方法允许在各种体系中研究结晶。为了准确地描述遗传算法的相互作用,需要从头计算描述,但运行第一性原理分子动力学(MD)是较为昂贵的。这个难题的解决方法是由Behler和Parrinello首先提出,即在 大量适当选择的构型集合 上 训练一个神经网络 (NN)。这意味着要精确地计算总能量和力,通常在密度泛函理论(DFT)水平上,并优化NN的参数,以便最好地重现这些数据。
近日,来自 西北工业大学 Haiyang Niu 、 苏黎世联邦理工学院Michele Parrinello 等研究者选择使用最近开发的MD的深度势 (DeePMD)方法,结果表明,所得到的 神经网络力场能够较好地描述合金的结构和相关性质。重要的是,计算了镓在较宽的温度和压力范围内的相图,与实验结果吻合较好。通过比较α-Ga和β-Ga的成核性能,其次发现,亚稳态β-Ga的形成在动力学上优于优于174 K以上的热力学稳定α-Ga。
在神经网络的训练中,对构型的仔细选择是神经网络潜能训练过程成功的关键。此文的重点是研究Ga相图及其成核过程,因此需要得到液体和固体的所有特征结构,最重要的是两相共存的成核区域的特征结构。在这里,研究者着手研究广义Ga相图时,其归纳了Bonati和Parrinello的方法,使用多热-多压系统的模拟来训练神经网络,从而为神经网络提供所有相关构型。神经网络训练过程如图2的四个步骤所示。这四个步骤包括多热-多气压模拟,收集一套相关构型的训练集,在DFT水平上计算能量和力以及训练神经网络势。该程序是基于三种方法的结合,即
多热-多压模拟
,
选择有效的集体变量
(CVs)和
使用DeePMD方法
。通过迭代这个过程,研究者能够获得DFT的优质势。
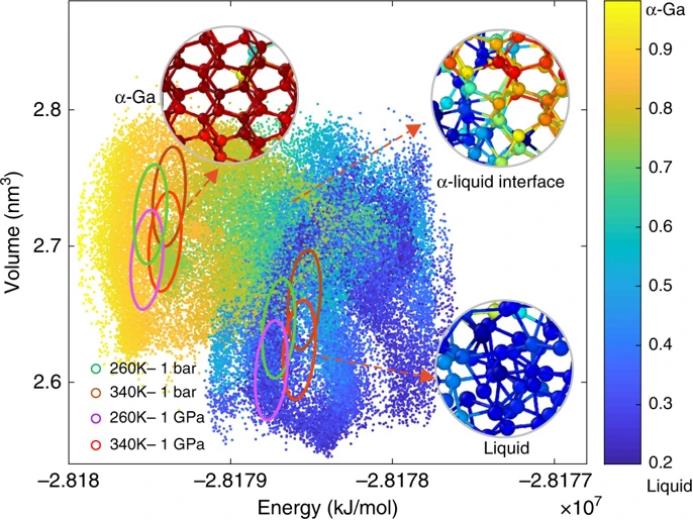
值得注意的是,使用
多热-多压模拟
是研究者方法的一个关键特征,它允许研究者建立一个
可靠的神经网络势
,进而确定
遗传算法相图
。在这种模拟中,研究者探索了
大量的排列方式
,从
典型的液体Ga
到通过
中间态的α-Ga
。正如图3中报告的,与标准等温-等压模拟相比,这些构型涵盖了更大范围的能量和体积。将神经网络暴露于各种物理相关构型中,是训练一个在全温度压力区域有效的神经网络的关键。
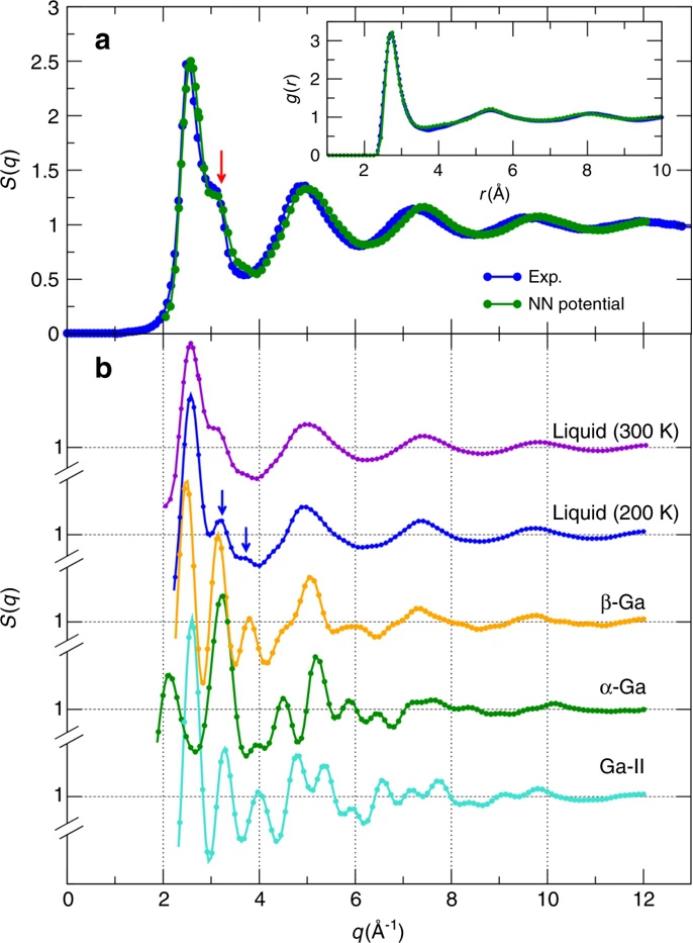
如图4所示,NN力场得到的液体镓的
径向分布函数g(r)
和
静态结构因子S(q)
,与
实验结果一致
。将液态Ga的g(r)积分到第一个最小值,得到的平均配位数为11.5,这与结构致密的液态金属相似,说明其原子环境的各向同性程度高于α-Ga。这些特征可能会导致密度异常,在冻结成α-Ga时,密度会膨胀~3.1%左右。此外,在S(q)的高-q侧观察到一个肩,与实验观察结果非常一致。研究者还比较了液体Ga与α-,β-以及Ga-II的S(q)。结果表明,如图4b所示,β-Ga与液态Ga的S(q)的峰位置基本一致,表明
β-Ga与液态Ga
之间存在潜在的结构相似性
。
另一方面,可以观察到在
α-镓与液相Ga之间的显著差异
,与之前的工作报告的结果一致。虽然Ga-II的S(q)与液体镓的S(q)也有一定的相似性,但在这种情况下Ga-II的作用并不重要,因为在环境温度和压力下Ga-II是高度不稳定的。研究者还计算了不同温度和压力下液体镓的g(r)和S(q)。结果表明,随着
温度的降低和压力的增大
,
液态镓的结构变得更加明显
。
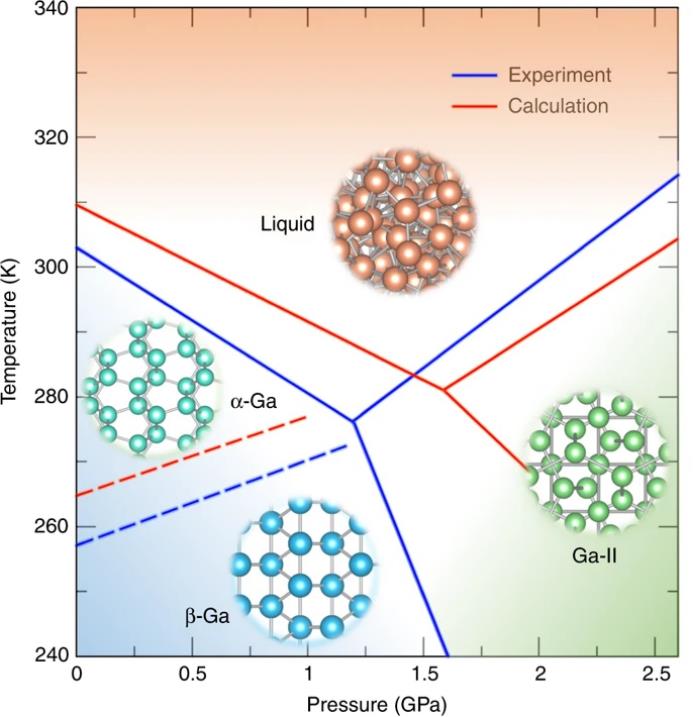
在这里,研究者目标是计算在240-340 K和0-2.6 GPa范围内的相图。为了实现这个目标,研究者进行了三次多热-多压模拟。三次多热-多压模拟得到的
镓相图
如图5所示,与
实验数据一致
。三相交叉点液-α-II位于280.9 K和1.58 GPa,与实验的276.2 K和1.19 GPa相当。研究者在表1中总结了这里
所研究的固相的熔点温度和晶格参数
,熔融温度和晶格参数
与实验结果基本一致
。
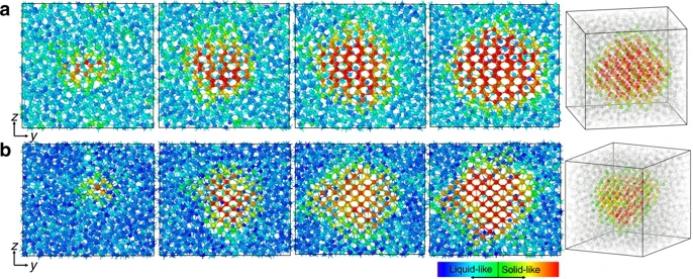
研究者为了研究固相的成核行为,使用神经网络势进行了多次元动态模拟。图6显示了来自于α-Ga和β-Ga均匀形核过程的快照。
在这两种情况下都可以观察到类似的特征
。最初,晶体胚胎在液相中随机形成。最终,一个团簇占据了主导地位,并稳定地成长为一个大晶体。有趣的是,在y-z平面上,
α-镓的晶体团看起来是相对球形的
,而
β-镓的晶体团是有棱角的平行四边形
。从三维角度来看,两种情况下的原子核都接近于球体,如图6所示。
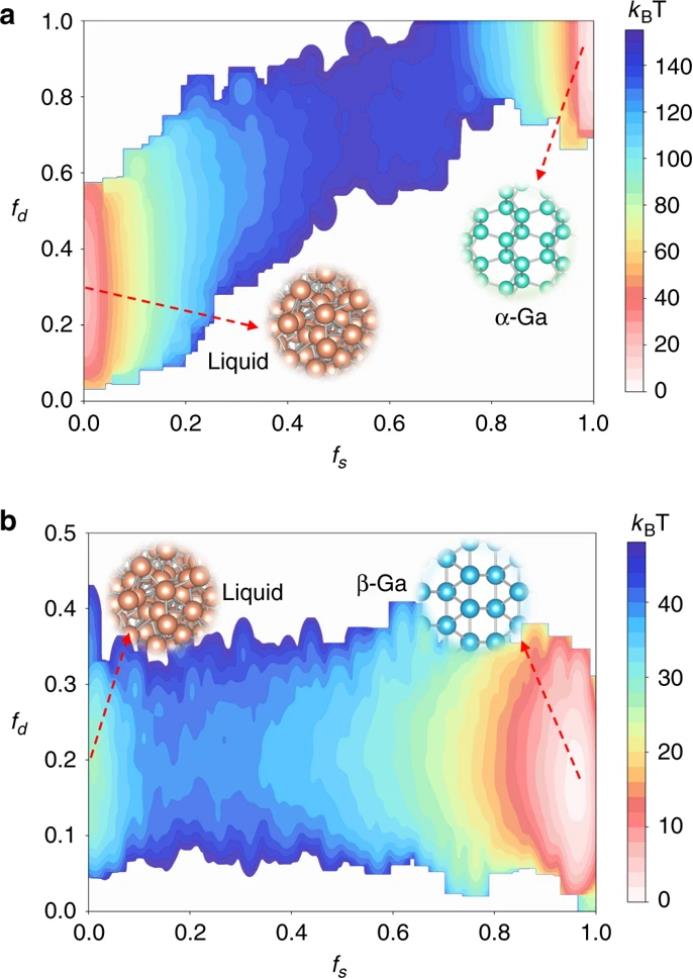
另一个潜在的问题是成核过程中的α-镓中的Ga
2 二聚体特征是如何演变的。为了解决这个问题,研究者计算了自由能表面(FES)作为一个函数,在280 K()和1 bar下,系统中有144个Ga原子的固相原子f
s 和Ga
2 二聚体f
d 的分数。如图7a所示,研究者的CVs很好地描述了在α-Ga和液体Ga之间的转变。在固相中,f
s 和f
d 都等于1,其中液相中f
s 和f
d 分别约等于0和0.25。
两个盆地大致呈线性连接,表明
Ga
2 二聚体与类固相原子的形成有很强的相关性
。在液相中,Ga
2 二聚体的取向是随机的,沿成核方向的二聚体有利于形成固相原子。相比之下,在具有144个原子的体系中,β-Ga在240 K()和1 bar条件下成核,FES作为类固相原子的比例f
s 和Ga
2 二聚体f
d 的函数如图7b所示。由于热波动,在β-Ga中也存在少量二聚体。由于液体Ga与β-Ga的结构相似,在β-Ga成核过程中f
d 没有明显变化。这些模拟阐明了Ga
2 二聚体在α-Ga成核过程中的演变。然而,由于严重的有限尺寸效应,不能定量地比较α-Ga和β-Ga的成核能垒。
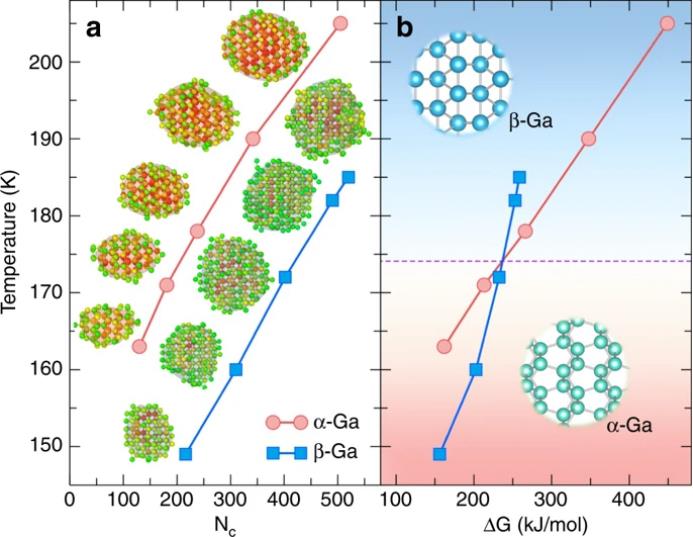
在CNT成立的假设下,播种技术可用于获得临界过冷核尺寸和成核能势垒。通常,将球状团簇插入本体液体中来创建初始模拟系统。这里,我们使用从meta动力学模拟中提取的构型(图6)作为种子模拟的起点,改进了标准程序。然而,在目标温度下,从α-Ga和β-Ga的成核中提取的晶型在~0.2 ns左右。研究者为每个阶段选择了5个初始构型,然后在不同的温度下执行MD运行,并监视集群大小,以确定每个集群的关键温度。
在图8中,研究者报告了α-Ga和β-Ga在不同温度下的临界核尺寸N
c 和成核势垒。
首先可以看出,在相同温度下,β-Ga的临界尺寸要比α-Ga的临界尺寸大得多。此外,在173 ± 3 K温度下,两晶相的成核势垒曲线相交。在此温度以上,β-Ga的成核能势垒低于α-Ga的成核能势垒,说明过冷液Ga有利于β-Ga成核,而不利于α-Ga成核。该结果阐述了实验结果,
液体
Ga的结晶在微米或亚微米尺寸下不会形成α-Ga,而主要是β-Ga
。
此外,研究者还比较这两种情况下的界面自由能γ,结果表明,β-Ga的界面能比α-Ga的界面能小约3倍。研究者还估算了在170 K和180 K时,
α-Ga和β-Ga的成核速率
J 分别为~ 2.4×10
- 23 和5.3×10
- 31 m
- 3 s
- 1 。换句话说,平均要等~4.8×10
17 天和~2.2×10
25 天,才能在170 K和180 K的1 m
3 液体Ga中看到单个核的形成。这表明,在
这些条件下不会发生均相形核
,而
非均相形核是正常的结晶机制
,这与
实验结果一致
。该结果同时也证明了为什么液体Ga可以在成核开始之前被实验过冷到极低的温度。
综上所述,研究者结合了众多先进的计算技术,为具有许多复杂的键合和结构性质的镓构建一个神经网络力场。结果表明,所得到的神经网络力场不仅能
很好地描述液态镓的结构
,而且能
很好地反映所研究的固相
(如:α-Ga、β-Ga、Ga-II)的性质。这些性质包括熔化温度、点阵参数和熔化焓。此外,还得到了镓在较宽的温度和压力范围内的相图,与实验结果很好地吻合。该项研究为计算相图和研究复杂材料的形核提供了一种方法,以
合理的成本进行从头计算
。
Niu, H., Bonati, L., Piaggi, P.M. et al. Ab initio phase diagram and nucleation of gallium. Nat Commun 11 , 2654 (2020). https://doi.org/10.1038/s41467-020-16372-9
DOI:https://doi.org/10.1038/s41467-020-16372-9
原文链接:https://www.nature.com/articles/s41467-020-16372-9
做计算,学计算,请认准唯理计算
——你身边更值得信赖的计算团队
唯理计算可以提供计算服务、培训课程、超算租赁等,有需要的小伙伴,可以联系我们:
做计算,学计算,就找唯理计算,唯理计算和您在一起!
小福利:
17812574221
备注:模拟计算进群
群里可以和老师一起探讨问题,老师也会帮助解答问题的哦~
以上是关于文献速递:神经网络算法&计算化学中从头算镓的成核与相图的主要内容,如果未能解决你的问题,请参考以下文章
文献速递 | 基于内窥镜图像的卷积神经网络在幽门螺杆菌感染诊断中的应用
本周最新文献速递20210822
电影脚本的多层网络模型 | 网络科学论文速递10篇
文献速递20200524
文献速递20200524
CASPT2是量子化学的啥计算方法,中文名叫啥?