生物信息学的发展与未来
Posted Bioinfor 生信云
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了生物信息学的发展与未来相关的知识,希望对你有一定的参考价值。
欢迎关注bioinfor 生信云!有一起想做公众号的朋友欢迎联系我!
1.发展历史
今天我们大部分人认为,现代生物信息学是最近出现的,有利于下一代测序数据分析。然而,生物信息学的起源发生在50多年前,当时台式计算机仍然是一个假设,DNA还不能测序。生物信息学的基础是在20世纪60年代初随着计算方法在蛋白质序列分析中的应用(特别是从头序列组装,生物序列数据库和替代模型)而奠定的。后来,DNA分析测序也出现了,因为分子生物学方法的平行进展,这使得DNA及其测序更容易操作,以及计算机科学兴起,以及更适合处理生物信息学任务的新型软件。在 20 世纪 90 年代到 2000 年代,测序技术的重大改进以及成本的降低导致数据呈指数级增长。“大数据”的到来在数据挖掘和管理方面提出了新的挑战,需要从计算机科学到该领域的更多专业知识。再加上生物信息学工具数量的不断增加,生物大数据对生物信息学结果的预测能力和可重复性具有深远的影响。最近的子学科,如合成生物学,系统生物学和细胞建模,已经从计算机科学和生物学之间不断增长的互补性中出现[1]。
1.1人类基因组计划
人类基因组计划(HGP)是历史上最伟大的探索成就之一。HGP不是对行星或宇宙的向外探索,而是由一个国际研究小组领导的向内发现之旅,旨在对我们人类的所有基因(统称为基因组)进行测序和绘制图谱。从1990年10月1日开始,到2003年4月完成,HGP让我们第一次有能力阅读完整的人类基因蓝图。最终,它提供了一个非常高质量的人类基因组序列,几乎完成,占人类基因组的92%。当时,DNA测序技术无法继续完成这项任务。剩余的约8%的人类基因组特别难以测序,因为这些区域含有高度重复的DNA。自人类基因组计划结束以来,更强大的技术,如长期读取的DNA测序方法和更新的计算工具,帮助研究人员对重复丰富的基因组区域进行测序。
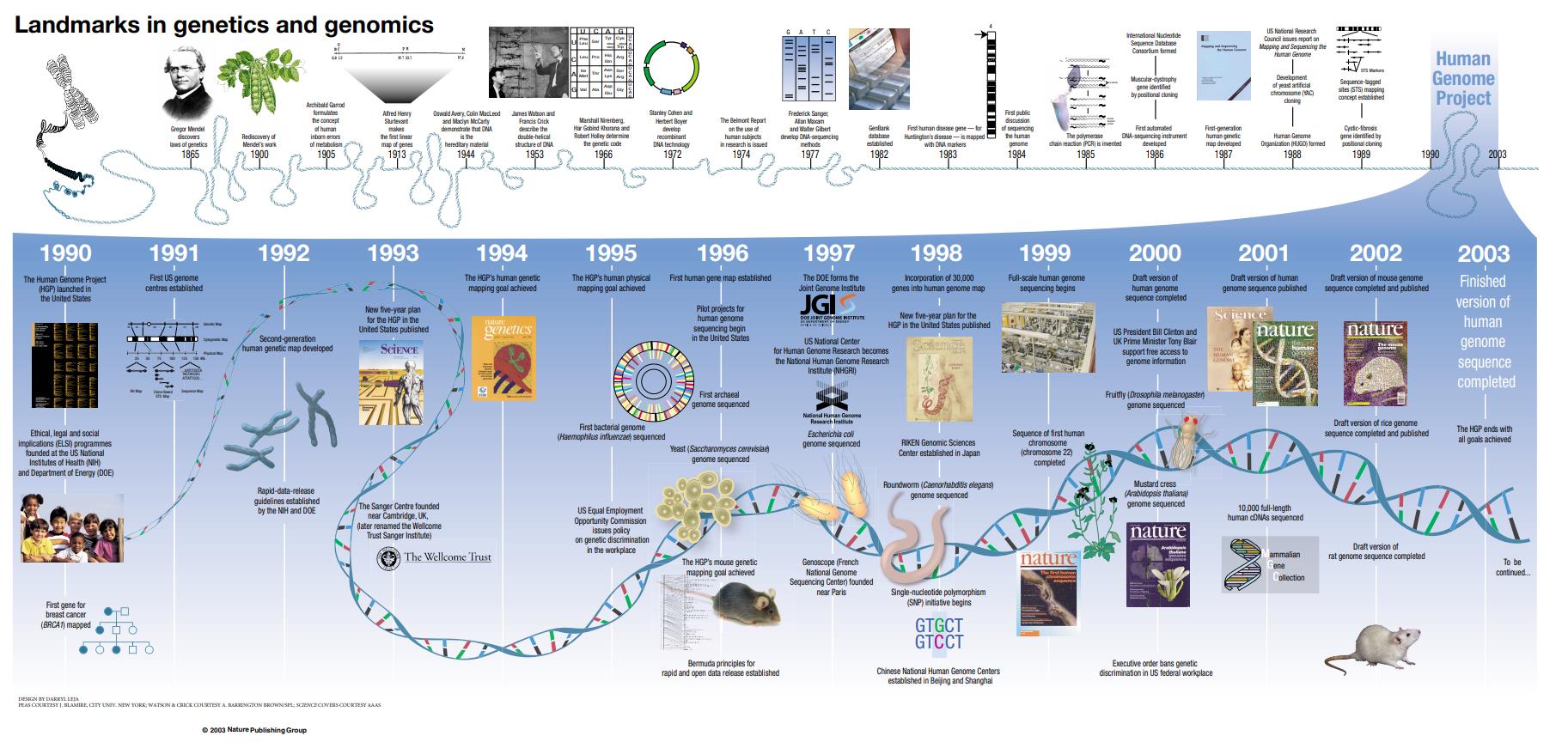
Nature volume 422, pages835–847 (2003)
2022年4月1日,《科学》(Science)杂志一口气发表6篇来自端粒到端粒(T2T)联盟论文,公布历史首个完整无间隙的人类基因组序列,报告了人类基因组的第一个真正完整的序列。该序列超过30亿个碱基对长,横跨23条染色体是完全无间隙的。T2T联盟进一步使用这个新完成的基因组序列作为参考,发现了超过200万个额外的基因组变异。这些信息对于全面了解人类基因组如何变化以及调查这些新发现的变异如何影响健康和疾病是有价值的[2-7]。
2 发展目标
生物信息学是计算科学的一个领域,与生物分子序列的分析有关。通常是指基因,DNA,RNA或蛋白质,并且在比较生物体内或生物体之间的蛋白质和其他序列中的基因和其他序列,观察生物体之间的进化关系以及使用存在于DNA和蛋白质序列中的模式来弄清楚它们的功能特别有用。你可以把生物信息学看作是遗传学的语言学部分。也就是说,语言学的人正在研究语言中的模式,这就是生物信息学人们所做的——在DNA或蛋白质序列中寻找模式。
为了研究正常细胞活动在不同疾病状态下是如何改变的,必须结合生物数据来形成这些活动的全面图景。因此,生物信息学领域已经发展到现在最紧迫的任务涉及对各种类型数据的分析和解释。这还包括核苷酸和氨基酸序列,蛋白质结构域和蛋白质结构。解释数据的实际过程被称为计算生物学。生物信息学和计算生物学中重要的子学科包括:
开发和实施计算机程序,以便能够有效地访问,管理和使用各种类型的信息。开发新算法(数学公式)和统计措施,以评估大型数据集成员之间的关系。例如,有一些方法可以在序列中定位基因,预测蛋白质结构和/或功能,并将蛋白质序列聚类为相关序列家族。
生物信息学的主要目标是增加对生物过程的理解。然而,它与其他方法的不同之处在于,它专注于开发和应用计算密集型技术来实现这一目标。示例包括:模式识别、数据挖掘、机器学习算法和可视化。该领域的主要研究工作包括序列比对,基因发现,基因组组装,药物设计,药物发现,蛋白质结构比对,蛋白质结构预测,基因表达和蛋白质 - 蛋白质相互作用的预测,全基因组关联研究,进化建模和细胞分裂/有丝分裂。
生物信息学现在需要创建和改进数据库,算法,计算和统计技术以及理论,以解决生物数据管理和分析中出现的形式和实际问题。
在过去的几十年里,基因组和其他分子研究技术的快速发展与信息技术的发展相结合,产生了大量与分子生物学相关的信息。生物信息学是这些数学和计算方法的名称,用于收集对生物过程的理解。
生物信息学中的常见活动包括绘制和分析DNA和蛋白质序列,对齐DNA和蛋白质序列以进行比较,以及创建和查看蛋白质结构的3D模型。
https://en.wikipedia.org/wiki/Bioinformatics
2.1不同的研究水平
1.能够简单的运行程序/脚本
2.能够将程序/脚本改进之后运用到自己的项目
3.能够独立的写一个项目的程序/脚本
程序员 vs 科研工作者
程序员: 能够通过改进算法/优化程序/重写软件来设计性能最佳的程序
科研工作者: 能够选择最佳的分析策略来回答科学问题
3基因测序
自从1977年第一个噬菌体Phage Φ-X174 被测序以来,到目前为止已有数千种生物完成测序并储存在数据库中,对测序序列进行数据分析确定编码蛋白质,CDS,内含子,外显子等等。一个物种内或不同物种之间基因的比较可以探究蛋白质功能之间的相似性,或物种之间的关系。
3.1测序发展
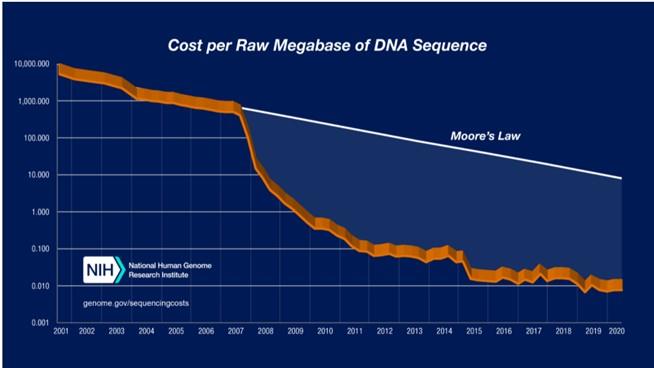
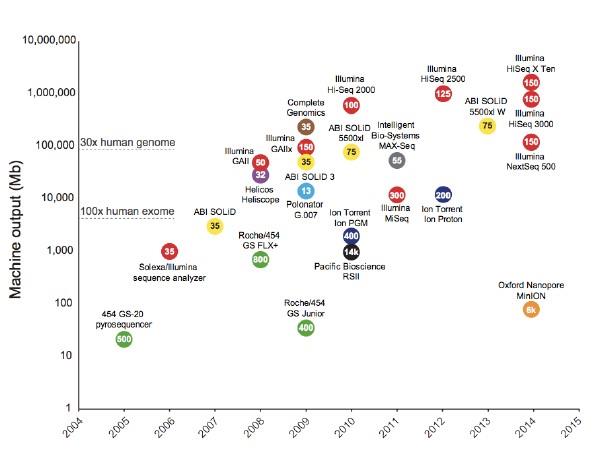
www.genome.gov/sequencingcostsdata
在这两个图表中,2001年至2007年10月的数据代表了使用基于Sanger法测序产生的成本(“第一代”测序平台)。从2008年1月开始,这些数据代表了使用“第二代”测序平台产生的成本。仪器的变化代表了近年来DNA测序技术的快速发展。
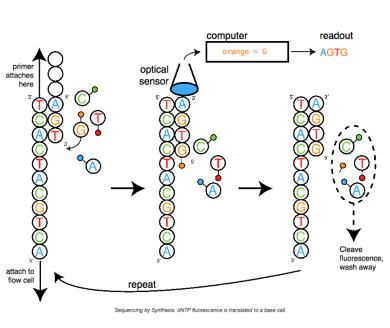
3.2基因测序
现在主流的测序平台包括Illumina、PacBio、Nanopore,前两种是边和成边测序通过荧光标记来识别不同的碱基,Nanopore是基于DNA分子通过单分子纳米孔时由于不同碱基的电阻不一样,膜两侧的电压不一样最终通过不同强度的电流信号来判断不同的碱基。
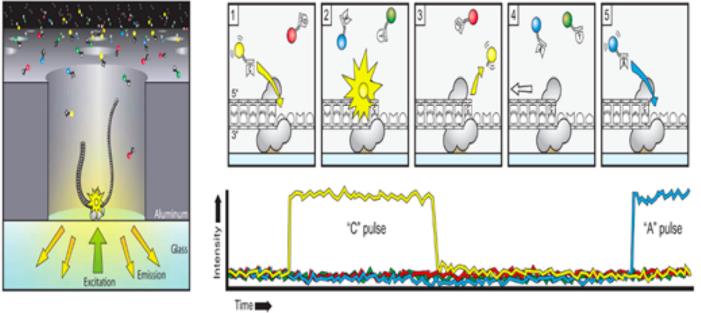
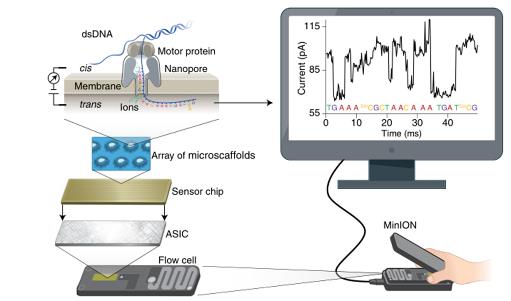
3.3基因组组装
在生物信息学中,基因组组装代表了将大量短DNA序列重新组合在一起以重建DNA起源的原始染色体的过程。序列组装是执行下一代测序、PacBio SMRT测序或纳米孔测序后的基本步骤之一。已建立的基因组组装可以提交到数据库,例如EMBI,NCBI和Ensembl。有两种不同类型的基因组组装:从头组装和映射到参考基因组。从头组装是指在没有参考基因组的情况下下从头开始组装新的基因组。映射到参考基因组是指直接将清洗后的数据比对到参考基因组(物种的代表基因组)上,基因组组装变得更加容易,更快,更准确。
3.4基因组注释
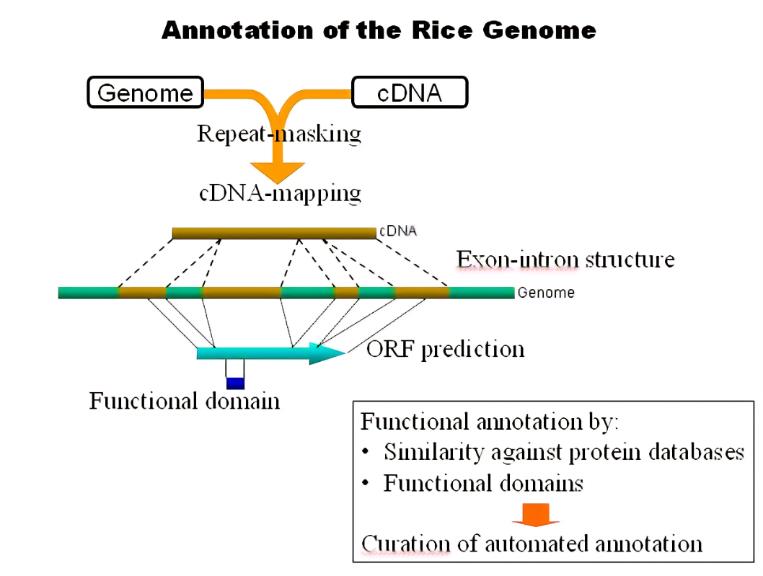
当我们做完基因组组装之后,我们接下来需要对我们的基因组注释,鉴定基因组元素及其功能的过程称为基因组注释,虽然“基因组注释”主要用于狭义上基因组上的基因结构(mRNA),但它最近已被用于广义上的任何基因组元素。在扩大注释范围后,我们有了关于各种其他功能元素的信息,包括非编码RNA,启动子和增强子序列,DNA甲基化位点等。尽管如此,基因组注释的核心特征仍然是基因列表,特别是蛋白质编码基因[8]。
3.5基因功能预测
通过计算方法进行基因预测以找到蛋白质编码区域的位置是生物信息学中的基本问题之一。基因预测基本上意味着沿着基因组定位基因。也称为基因发现,它是指识别编码基因的基因组DNA区域的过程。这包括蛋白质编码基因,RNA基因和其他功能元素,如调节基因。
其基础是发现EST(表达的序列标签),蛋白质或其他基因组与输入基因组之间的基因序列相似性。一旦某个基因组区域与EST,DNA或蛋白质之间存在相似性,就可用于推断该区域的基因结构或功能[9],常用的软件是BLAST。目前,大量的基因功能预测方法使用基因本体论(GO)作为功能分类的来源或结果来确认。
##4.基因组学是生命科学的基础
自从人类基因组计划完成之后,基因组学就成了我们生命科学的基础。就像我们建放房子先修地基一样,地基修好了我们才能不断的去加楼层,在人类基因计划之后又出现了计算基因组学、功能基因组学、比较基因组学、结构基因组学、宏基因组学、营养基因组学等等,这个房子以后会搭的越来越好看。
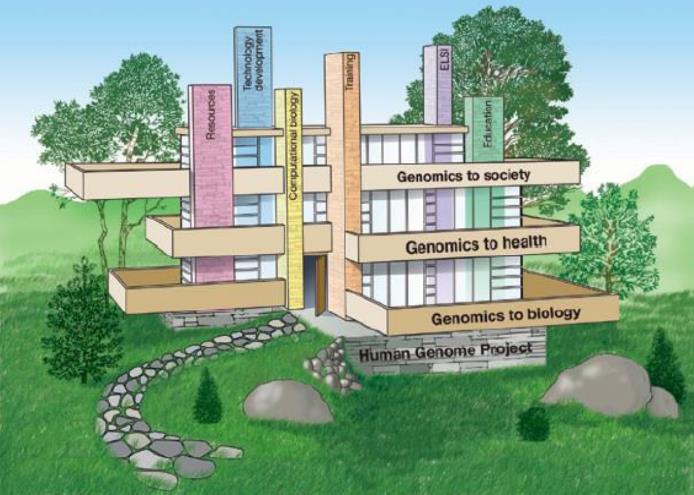
4.1计算基因组学
计算基因组学是指使用计算和统计分析从基因组序列和相关数据中破译生物学[10],包括DNA和RNA序列以及其他“后基因组”数据,结合计算和统计方法来理解基因的功能和统计关联分析,该领域通常也被称为计算和统计遗传学/基因组学。因此,计算基因组学可以被视为生物信息学和计算生物学的一个子集,但重点是使用全基因组(而不是单个基因)来理解物种的DNA如何在分子水平及其他水平上控制其生物学的原理。随着目前大量生物数据集的丰富,计算研究已成为生物发现最重要的手段之一。
4.2功能基因组学
功能基因组学(Functional genomics),功能基因组学是研究基因组的基因和基因间区域如何促成不同的生物过程。功能基因组学侧重于基因产物在特定环境中的动态表达,例如,在特定发育阶段或疾病期间。
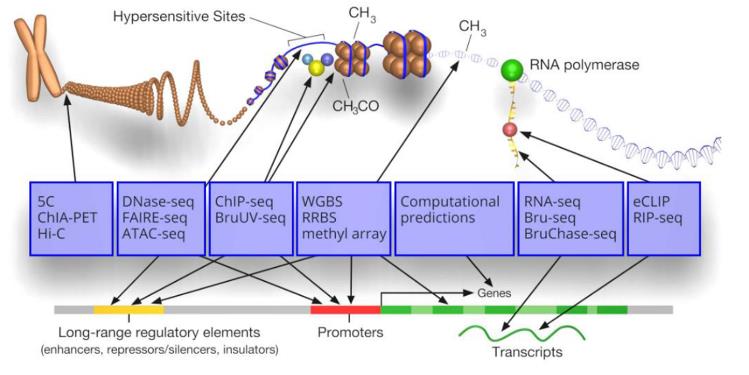
https://www.encodeproject.org/
4.3比较基因组学
比较基因组学(Comparative genomics)是基于基因组图谱和测序技术,对已知的基因特征和基因组结构进行比较以了解基因的功能、表达机制和不同物种亲缘关系的生物学研究。基因组的特征可包括的DNA序列,基因,基因顺序,调控序列,和其它的基因组结构标志。通过对不同亲缘关系物种的基因组序列进行比较,能够鉴定出编码序列、非编码调控序列及给定物种独有的序列。而基因组范围之内的序列比对,可以了解不同物种在核苷酸组成、共线性关系和基因顺序方面的异同,进而得到基因分析预测与定位、生物系统进化关系等方面的信息。
4.4结构基因组学
结构基因组学是一门用结构生物学方法研究整个生物体、整个细胞或整个基因组中所有的蛋白质和相关蛋白质复合物的三维结构的学科。主要利用实验方式(X射线晶体学、核磁共振波谱法和电子显微)来测定蛋白质结构,同时结合同源建模这一计算方式来推测蛋白质结构,和传统结构生物学不同的是,利用结构基因组学所测定的蛋白质结构通常是功能未知的蛋白质,通过三维结构信息来预测蛋白质功能。结构基因组学重视快速、高通量的蛋白质结构测定。包括三个重要的计划,蛋白质结构启动计划(Protein Structure Initiative, PSI),欧洲结构蛋白质组计划(Structural Proteomics in Europe, SPINE),中国结构基因组计划。
4.5宏基因组学
微生物在人体的食物消化、机体免疫等方面发挥着重要作用。在大多数情况下,微生物通过群落而非单一个体来发挥这些重要功能。水体、土壤、肠道和很多的人工生物环境(如废水处理、食品发酵、堆肥、沼气池等等)都具有很复杂的微生物群落,这些微生物相互作用、共同协作,一起完成复杂的代谢功能。环境样品中的微生物组成的群落构成了一个巨大而复杂的基因库,在这个基因库中既包含代表不同微生物身份的系统发育标记基因(如16S rRNA基因),也包含各种代谢功能基因,它们统称为宏基因组(metagenomics,又称宏基因组、环境基因组或生态基因组),这些基因确定了样品微生物群落的组成与功能,研究样品的基因组是认识复杂微生物群落的主要途径。
宏基因组学在开发微生物资源多样性、筛选获得新型活性物质、发掘与抗生素抗性、维生素合成及污染物降解相关的蛋白质等方面展示了很大的潜力。
人类微生物组计划(Human Microbiome Project,HMP)是美国国立卫生研究院(NIH)于2008年发起的一项旨在鉴定与阐明和人类健康与疾病相关的微生物功能的计划。于2007年启动,第一阶段 (HMP1) 专注于识别和表征人类微生物群。 第二阶段被称为“综合人类微生物组计划 ”(iHMP),于 2014 年启动,旨在产生资源来表征微生物组并阐明微生物在健康和疾病状态中的作用。https://www.hmpdacc.org/
4.6营养基因组学
20世纪90年代人类基因组计划的启动以及随后的人类DNA测序图谱开创了“大科学时代”,启动了我们今天所知道的营养基因组学(Nutrigenomics)领域[11]。营养基因组学是研究食物如何与基因相互作用,并解释可能影响我们食物中维生素,矿物质和化合物需求的个体遗传差异。每个人都根据其基因组成以不同的方式吸收,代谢和运输化学物质,营养基因组学开启了个体营养图谱的蓝图。
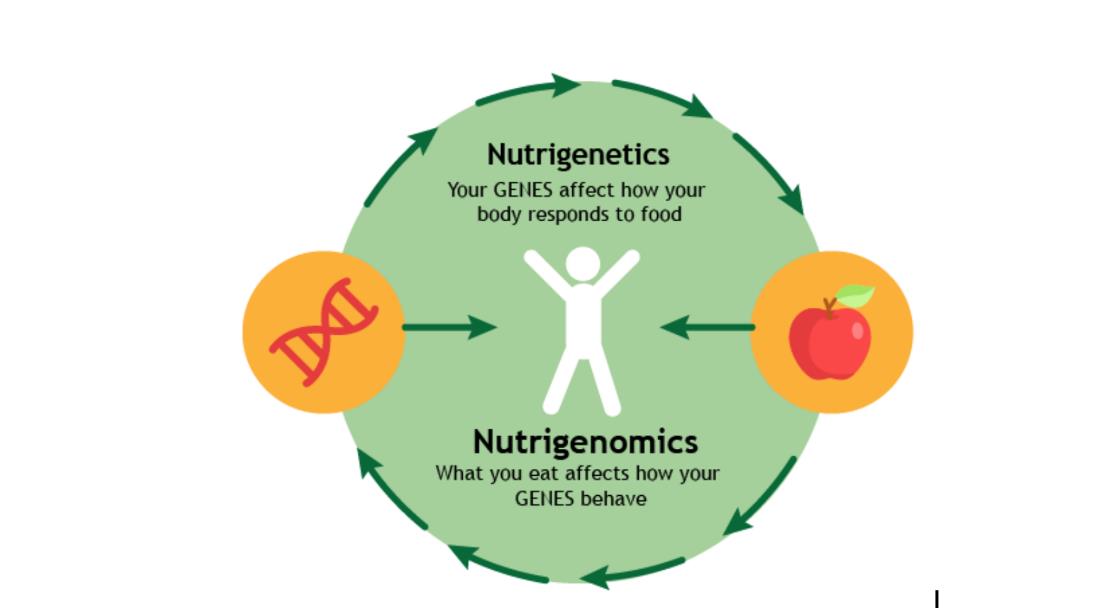
4.7代谢组学
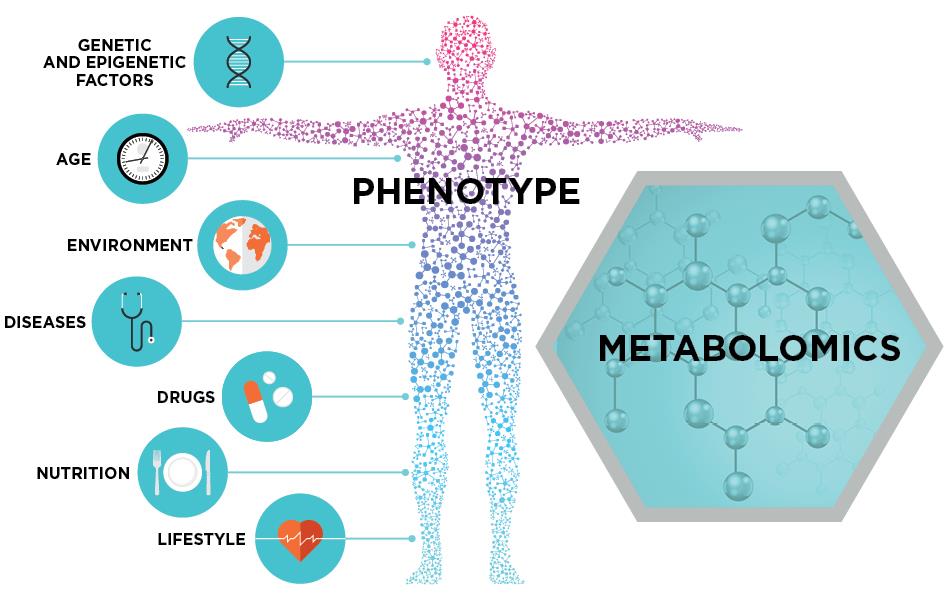
代谢物是在代谢过程中化学转化的小分子,因此,它们提供细胞状态的功能参数。与功能分别受表观遗传调节和翻译后修饰影响的基因和蛋白质不同,代谢物是生物活性的直接特征,因此它们更容易与表型相关联。代谢组是生物途径的输入和输出的量度,因此,通常被认为比其他组学(如基因组学或蛋白质组学)更能代表细胞的功能状态。此外,许多代谢物在各种动物物种中都是保守的,有助于将实验动物的研究结果外推到人类。测量代谢组的常用技术包括质谱(MS)和核磁共振波谱(NMR),它可以检测数百到数千种独特的化学实体。在这种情况下,代谢组学(metabolomics)已成为一种强大的方法,已被广泛用于临床诊断[12]。
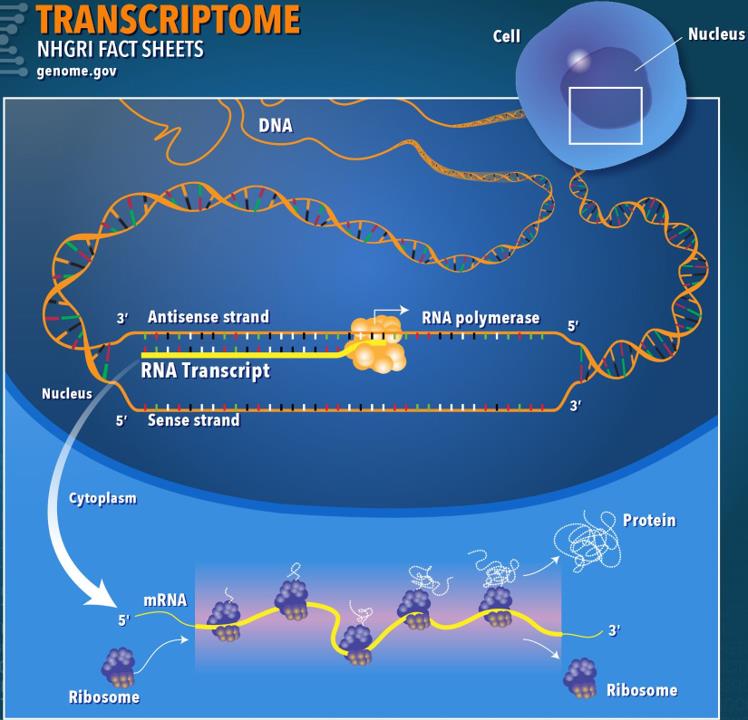
4.8转录组学
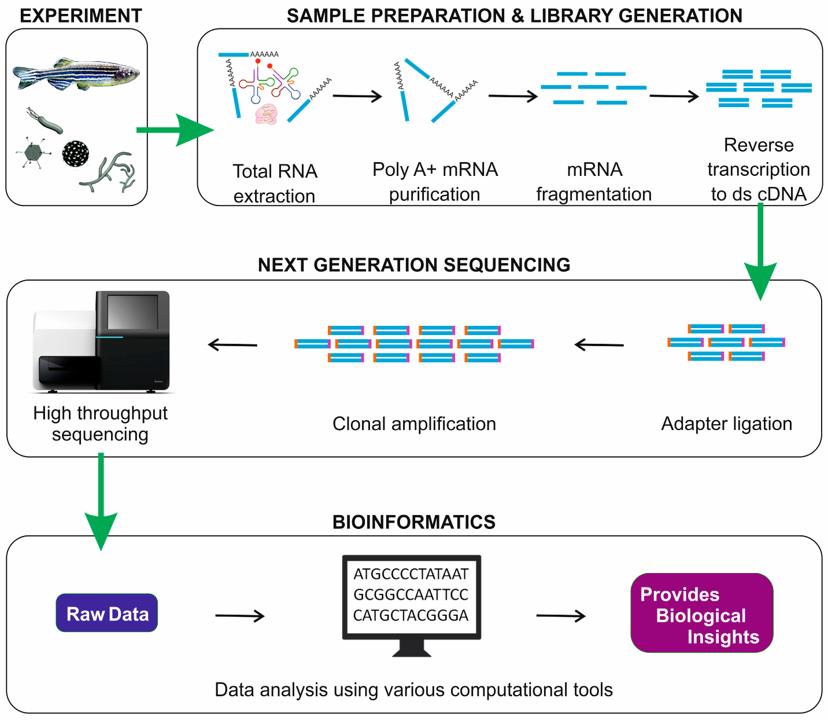
转录组学(Transcriptomics)是对基因型在给定时间产生的RNA转录本的分析,该转录本在基因组,蛋白质组和细胞表型之间提供了联系。转录组是所有RNA分子的集合,包括mRNA,rRNA,tRNA和在一个或一个细胞群中产生的非编码RNA。转录组学也称为表达谱分析,检查给定细胞群中RNA的表达水平[13]。
RNA-Seq一种使用深度测序技术的转录组分析方法,它使用下一代测序(NGS)来揭示特定时间生物样品中RNA的存在和数量,分析不断变化的细胞转录组[14]。
4.9蛋白质组学
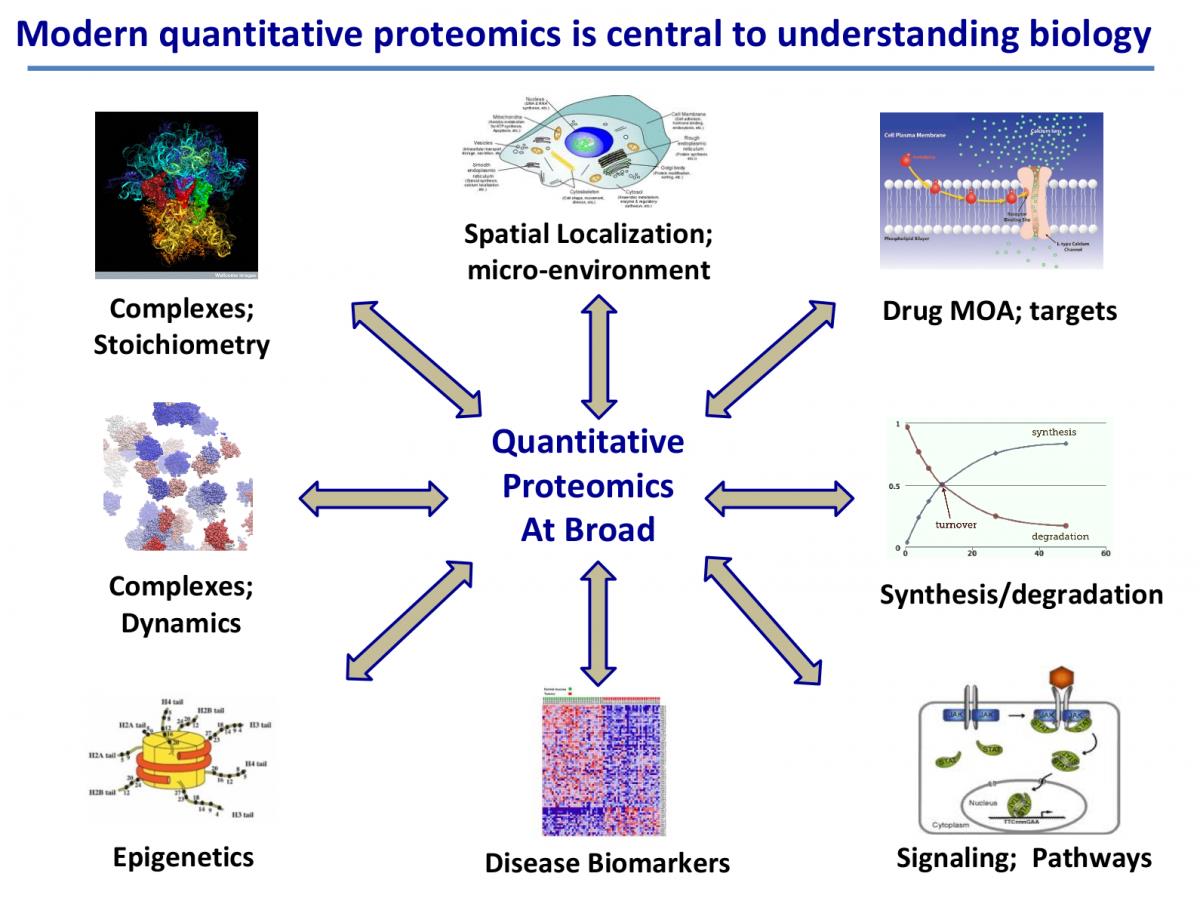
蛋白质组学(Proteomics)是对蛋白质特别是其结构和功能的大规模研究,是在90年代初期,由马克·威尔金斯(Marc Wikins)和学者们首先提出的新名词。它补充了其他“组学”技术,如基因组学和转录组学,以阐明生物体蛋白质特征,并识别特定蛋白质的结构和功能。它比基因组学更复杂,因为生物体的基因组或多或少是恒定的,而蛋白质组因细胞而异。蛋白质组学研究的关键技术包括质谱分析、X射线晶体学、核磁共振和凝胶电泳。基于不用蛋白质组学的技术用于不同的研究环境,例如检测各种诊断标志物,疫苗生产的候选物,了解致病机制,响应不同信号的表达模式的改变以及解释不同疾病中的功能蛋白途径[15]。
5.未来的发展方向
5.1多组学联合分析
单一的“组学”技术提供了构成细胞,组织和生物体的分子视图。然而,这种观点通常仅限于单个水平,如基因组,转录组,蛋白质组,代谢组等水平。集成单个级别以生成全局视图通常称为多组学方法。多组学方法对于理解癌症等复杂疾病非常有用,其中疾病病因受到多种遗传和环境因素的影响。多组学方法可以大致分为基于遗传,表型和环境因素的方法[16]。基于基因型的多组学方法旨在使用全基因组关联研究来鉴定与疾病风险相关的位点。进一步检查位点区域可以帮助确定可能在疾病启动中起作用的候选基因。通过探索基因组和转录组水平上的突变或表达变化,可以进一步验证相关基因。其次,基于表型的多组学方法探索了疾病,临床因素和基于组学的数据之间相关性的知识。第三,基于环境的多组学方法结合了来自组学数据(如微生物组,基因组或代谢组水平)的信息,并估计与吸烟和饮食等环境因素的关联。
多组学提供了一种更全面的方法来解决生物学问题,方法是使用来自不同平台的综合信息在不同维度上观察它。
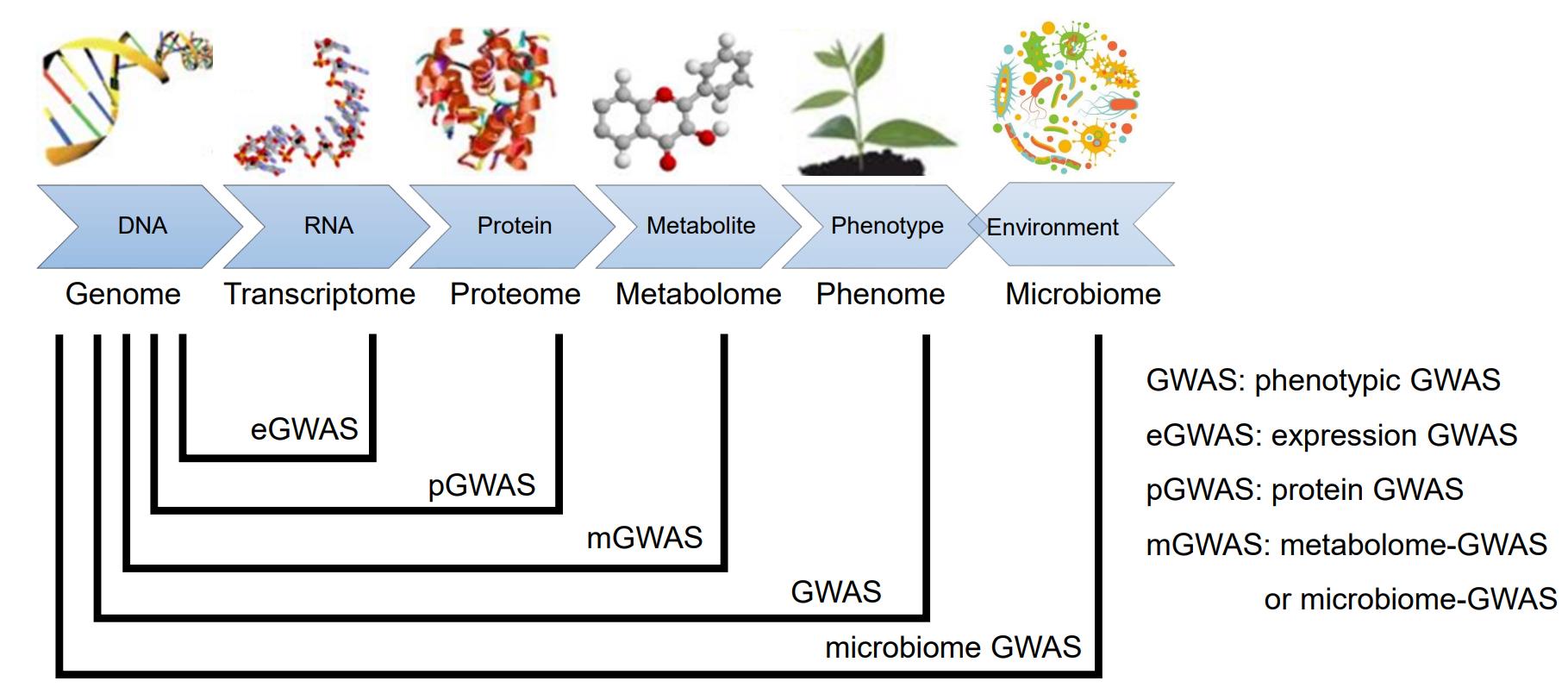
5.2单细胞转录组
RNA-seq通常是混合测序,数据代表了数千至数百万个细胞的基因表达模式的平均值;这可能会掩盖细胞之间的生物学相关差异。单细胞RNA-seq(scRNA-seq)代表了一种克服这个问题的方法。通过分离单个细胞,捕获其转录本并生成测序文库,其中转录本被映射到单个细胞,scRNA-seq能够以前所未有的分辨率评估细胞群和生物系统的基本生物学特性[17]。
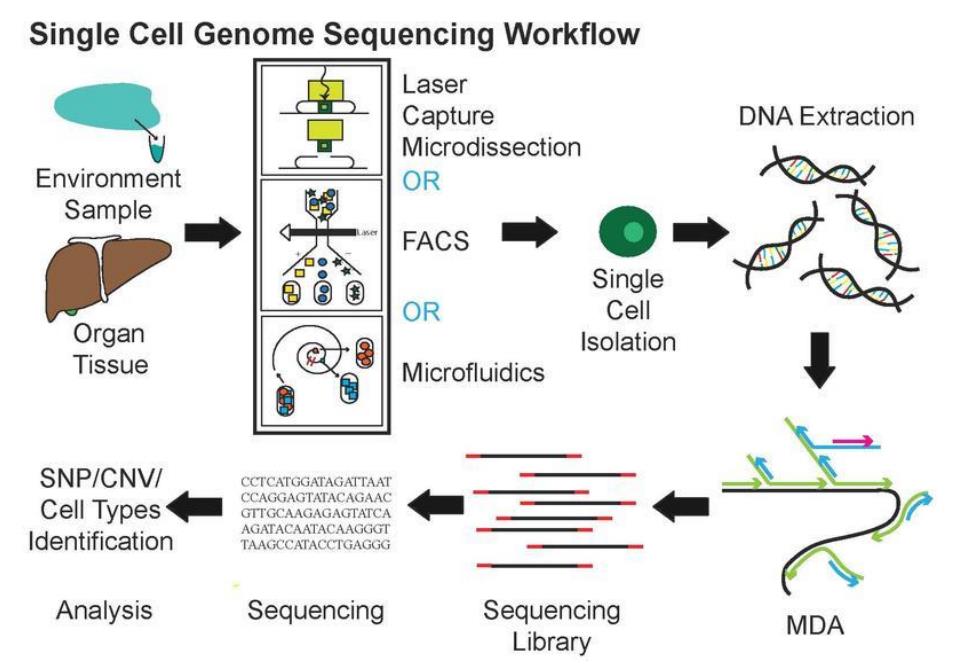
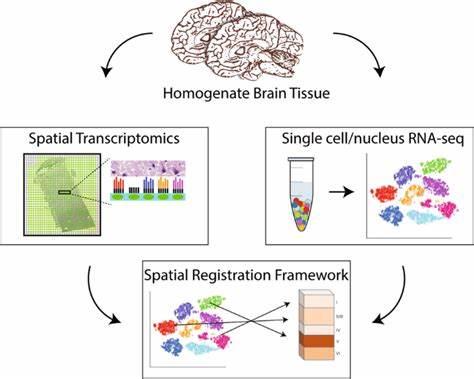
5.3空间转录组
空间转录组学是一系列方法的总体术语,旨在将细胞类型(由mRNA读数识别)分配到它们在组织学切片中的位置[18]。
空间转录组测序的工作流程主要分为两部分:组织学部分和组学部分。组织学部分包括样品包埋、切片、固定、染色和成像,并记录切片的形态学信息;组学部分包括cDNA合成、扩增、接合和测序,并记录该部分的转录本信息和空间位置信息。使用10X Genomics空间转录组测序技术,用于文库构建的每张载玻片都有四个捕获区域,其中每个捕获区域包含5000个条形码斑点,每个斑点具有唯一的条形码序列。组织部分的细胞会释放mRNA,迁移到每个斑点的mRNA会用相应的条形码序列标记,然后构建和测序文库。最后,根据数据的条形码信息对数据进行分析,确定哪些数据来自哪个位置,从而实现空间基因表达的可视化。
【参考文献】
[1] GAUTHIER J, VINCENT A T, CHARETTE S J, et al. A brief history of bioinformatics [J]. Brief Bioinform, 2019, 20(6): 1981-96.
[2] AGANEZOV S, YAN S M, SOTO D C, et al. A complete reference genome improves analysis of human genetic variation [J]. Science, 2022, 376(6588): eabl3533.
[3] NURK S, KOREN S, RHIE A, et al. The complete sequence of a human genome [J]. 2022, 376(6588): 44-53.
[4] ALTEMOSE N, LOGSDON G A, BZIKADZE A V, et al. Complete genomic and epigenetic maps of human centromeres [J]. Science, 2022, 376(6588): eabl4178.
[5] GERSHMAN A, SAURIA M E G, GUITART X, et al. Epigenetic patterns in a complete human genome [J]. Science, 2022, 376(6588): eabj5089.
[6] HOYT S J, STORER J M, HARTLEY G A, et al. From telomere to telomere: The transcriptional and epigenetic state of human repeat elements [J]. Science, 2022, 376(6588): eabk3112.
[7] VOLLGER M R, GUITART X, DISHUCK P C, et al. Segmental duplications and their variation in a complete human genome [J]. Science, 2022, 376(6588): eabj6965.
[8] SALZBERG S L. Next-generation genome annotation: we still struggle to get it right [J]. Genome biology, 2019, 20(1): 92.
[9] WANG Z, CHEN Y, LI Y. A brief review of computational gene prediction methods [J]. Genomics Proteomics Bioinformatics, 2004, 2(4): 216-21.
[10] KOONIN E V. Computational genomics [J]. Current biology : CB, 2001, 11(5): R155-8.
[11] MATHERS J C. Nutrigenomics in the modern era [J]. The Proceedings of the Nutrition Society, 2017, 76(3): 265-75.
[12] PATTI G J, YANES O, SIUZDAK G. Innovation: Metabolomics: the apogee of the omics trilogy [J]. Nature reviews Molecular cell biology, 2012, 13(4): 263-9.
[13] LOWE R, SHIRLEY N, BLEACKLEY M, et al. Transcriptomics technologies [J]. PLoS Comput Biol, 2017, 13(5): e1005457-e.
[14] WANG Z, GERSTEIN M, SNYDER M. RNA-Seq: a revolutionary tool for transcriptomics [J]. Nat Rev Genet, 2009, 10(1): 57-63.
[15] ASLAM B, BASIT M, NISAR M A, et al. Proteomics: Technologies and Their Applications [J]. Journal of Chromatographic Science, 2017, 55(2): 182-96.
[16] HASIN Y, SELDIN M, LUSIS A. Multi-omics approaches to disease [J]. Genome biology, 2017, 18(1): 83.
[17] OLSEN T K, BARYAWNO N. Introduction to Single-Cell RNA Sequencing [J]. Current protocols in molecular biology, 2018, 122(1): e57.
[18] STåHL P L, SALMéN F, VICKOVIC S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics [J]. Science, 2016, 353(6294): 78-82.
以上是关于生物信息学的发展与未来的主要内容,如果未能解决你的问题,请参考以下文章