【circRNA】circRNA的鉴定
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了【circRNA】circRNA的鉴定相关的知识,希望对你有一定的参考价值。
参考技术A
通过spliced reads的mapping能发现线性RNA和环状RNA的剪切方式不同。一个是正常的5’/3’前后剪切,一个是反向的5’/3’反向剪切(Memczak et al.2013.Nature)。
==== 建库策略 ====
环状RNA 测序数据量
建库策略的选择
所以,我们实验的方案都是采用环状RNA建库的。
==== 鉴定方法 ========
CircRNA检测的基本原理是去识别反向剪切的位点(back-splice),最主要的circRNA类型是外显子来源的,当然,在内含子、间区、UTR区域、lncRNA区域以及已知转录本的反义链区域也都鉴定到circRNA,同一个位点可能形成多个circRNA,每个circRNA可能包含一个或多个外显子。CircRNA的数量从几千到几万都有可能。要研究circRNA,鉴定是第一步,也是最重要的一步,目前已经有一些pipeline,鉴定得到的circRNA是否准确和全面,取决于算法的严谨性和可靠性。
根据已发表的文献,环状RNA的鉴定方法分为三类:
1. 从头预测(abinitio)的方法:find_circ(如下图)(Memczaketal., 2013),将不能和基因组比对上读段的两端各取20bp作为锚点,再将锚点作为独立的读段往基因组上比对并寻找唯一匹配位点,如果两个锚点的比对位置在线性上方向呈反向,那么就延长锚点的读段,直至找到环状RNA的接合位置(junction),若此时两侧的序列分别为GT/AG剪接信号,则判断为潜在的环状RNA。
2. 基于RNA-seq比对工具如:Tophat-fusion(KimandSalzberg, 2011)、Mapsplice(Wanget al., 2010)、STAR(Dobinet al., 2013)、segemehl(Hoffmannet al., 2014)等,以寻找融合基因的思想检测环状RNA(如下图):先将不能比对到转录本上的读段提取出来,再根据软件预测结果找出处于同一条染色体上的融合基因,最后根据基因组注释文件中外显子的边界来判断是否为环状RNA。(这也是目前最常用的方法)
3. 专门为寻找环状RNA而设计的算法和工具(如下图)如CIRI,它考虑了经典的环状RNA以及一些短外显子成环状RNA的情况,同样以GT-AG剪接信号和外显子边界得到环状RNA。
=== 鉴定方法比较 ====
2015,NAR发表了来自于丹麦奥尔胡斯大学(Aarhus University)的研究人员(Comparison of circular RNA prediction tools)利用普通的RNA-Seq数据比较了5种常用的环状RNA预测软件(见表1)。
这些算法都依赖外部比对工具,CIRCexplorer和Mapsplice需要有注释信息,其他三种可以不依赖注释信息,但是准确性会有所下降。耗用资源方面,仅finc_circ可以用单机运算(8G RAM),CIRI耗用资源最多。
测试数据:
物种:人
数据:SRR444655和SRR444975,未用RNaseR处理,该文章中主要用于分析的数据;
SRR444974和SRR445016,使用RNaseR处理,用于验证预测方法预测得到的circRNA准确性的数据。
测序仪器:Hiseq2000,pair-end。
测序量:31.4-41.3GB/样本。
预测结果比较
首先,研究人员用5个软件分别对同一个rRNA-depleted RNA-Seq数据集进行分析。他们发现各个算法给出的环状RNA数目从1500(circRNA_finder)到4000(CIRI)不等,并且只有854个同时被5个软件发现(如下图所示)。
为了验证软件给出的circRNA是否可信,研究人员试图引入线性RNA酶消化(RNase R)的RNA-Seq数据来判断预测到的circRNA是否存在假阳性。
结果显示不同的软件给出的circRNA对RNase R的抵制效率不同,其中,CIRI表现最差,有28.03%的假阳性率(见下图)。
研究人员还关心每个软件预测出的表达量最高的100个circRNA是否真的是环状。他们分别以junction read数目对环状RNA进行排序,观察表达量高的前100个环状RNA是否被线性RNA酶消化。
同样,在CIRI的预测中高表达的环状RNA有超过半数(63%)不可靠。MapSplice和CIRCexplorer是表现最好的两款软件,分别只有9%和6%的circRNA被消化(图下图)。
通过比较现有的circRNA预测软件,我们可以看到不同的算法表现差异较大,用户在使用的时候需要小心。(从venn图也可以看出其实overlap的概率是不高的)
CIRCexplorer和MapSplice输出最可信的circRNA列表,主要的原因是这两个算法依靠已知的基因注释文件,明确的序列注释信息可以帮助他们降低假阳性率,但也限制了这两个软件不能发现de novo的环状RNA。
CircRNA_finder和find_circ也有着很高的准确性,并且这两个软件可以独立于基因注释信息运行,预测全新的环状RNA。
由于单个软件往往在一个方面存在着一定的局限性,且数据表明能够被多个算法预测到的环状RNA有着较高的可信度,因此,在实际项目中,推荐大家多使用两到三个环状RNA预测软件,进而取它们的交集。
对于任意两种方法检测的效果,文中也做了比较:
一种ceRNA关系网络的新颖画法,R语言绘制冲击图(桑基图)教程
R语言绘制ceRNA冲击图(桑基图)
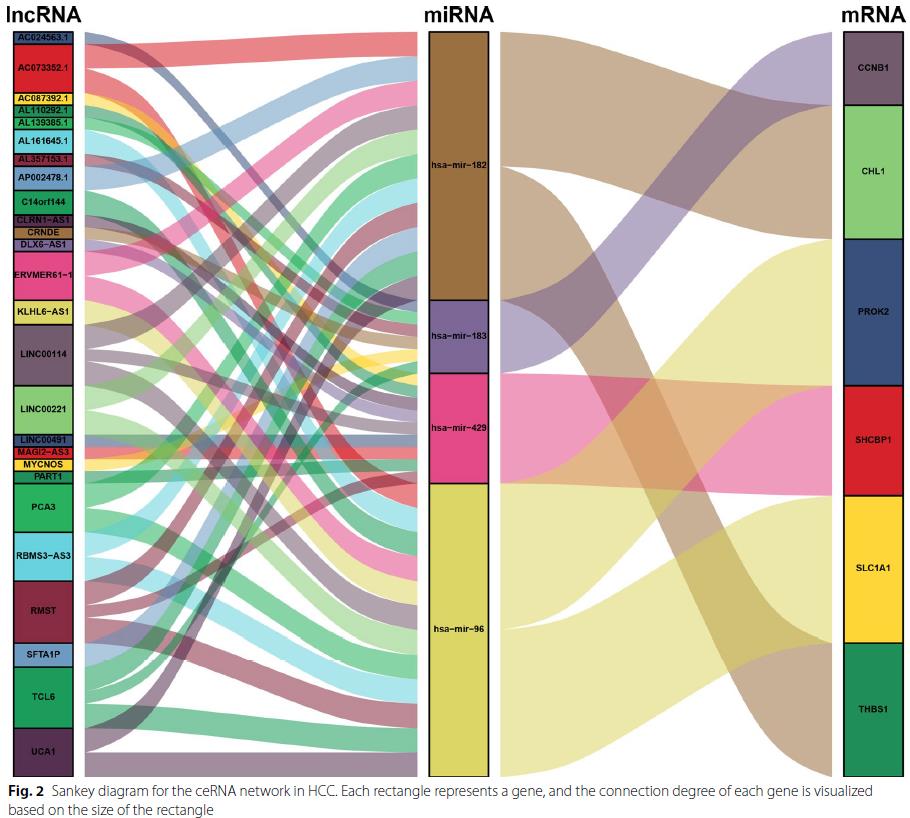

1 示例文件
示例文件一共两个,“lncRNA_miRNA.txt”记录了lncRNA和miRNA的靶向关系,“miRNA_mRNA.txt”记录了miRNA和mRNA的靶向关系。

2 对应靶向关系
首先将示例数据读入到R中,由于lncRNA-miRNA-mRNA的关系是分两张表记录的,需要将它们整合到一起,做个关系对应。
#读取数据,两个靶向关系表lncRNA_miRNA <- read.delim('lncRNA_miRNA.txt', sep = ' ', stringsAsFactors = FALSE)miRNA_mRNA <- read.delim('miRNA_mRNA.txt', sep = ' ', stringsAsFactors = FALSE)#整合靶向关系ceRNA <- merge(lncRNA_miRNA, miRNA_mRNA, by = 'miRNA')ceRNA$link <- 1ceRNA <- reshape::melt(ceRNA, id = 'link')variable <- summary(ceRNA$variable)ceRNA$flow <- rep(1:variable[1], length(variable))head(ceRNA) #查看整理后的数据结构

整理后的结构中:
variable指明为lncRNA、miRNA还是mRNA;
value为具体的分子名称;
flow为关系流,如果某lncRNA、miRNA和mRNA位于同一条靶向路径中,则flow中的数值是一样的。
3 R包ggalluvial的冲击图(桑基图)绘制
文件结构整理完毕后,绘制冲击图。
R语言中,绘制冲击图的R包其实有很多可以选择。考虑到ggalluvial包是示例文献中使用的作图R包,并且它延伸自ggplot2,语法结构和ggplot2是一致的比较简单易学,因此我们也以ggalluvial包的方法绘制冲击图。
#预指定颜色,lncRNA、miRNA 和 mRNA 总计 36 种,需指定 36 种颜色mycol <- c('#8DD3C7', '#FFFFB3', '#BEBADA', '#FB8072', '#80B1D3', '#FDB462','#B3DE69', '#FCCDE5', '#BC80BD', '#CCEBC5', '#FFED6F', '#E41A1C', '#377EB8','#4DAF4A', '#984EA3', '#FF7F00', '#FFFF33', '#A65628', '#F781BF', '#66C2A5','#6181BD', '#F34800', '#64A10E', '#FF00FF', '#c7475b', '#049a0b', '#BEAED4','#FDC086', '#FFFF99', '#386CB0', '#F0027F', '#4253ff', '#ff4308', '#D8D155','#64495D', '#7CC767')#ggalluvial 的冲击图library(ggalluvial)p <- ggplot(ceRNA, aes(x = variable, y = link,stratum = value, alluvium = flow, fill = value)) +geom_stratum() + #冲击图中的堆叠柱形图geom_flow(aes.flow = 'forward') + #冲击图连线绘制scale_fill_manual(values = mycol) + #颜色赋值geom_text(stat = 'stratum', infer.label = TRUE, size = 2.5) + #添加 lncRNA、miRNA 和 mRNA 标签scale_x_discrete(limits = c('lncRNA', 'miRNA', 'mRNA')) + #定义 lncRNA、miRNA 和 mRNA 列的展示顺序labs(x = '', y = '') + #去除 x 轴和 y 轴标题theme(legend.position = 'none', panel.background = element_blank(),line = element_blank(), axis.text.y = element_blank()) #去除背景和图例p
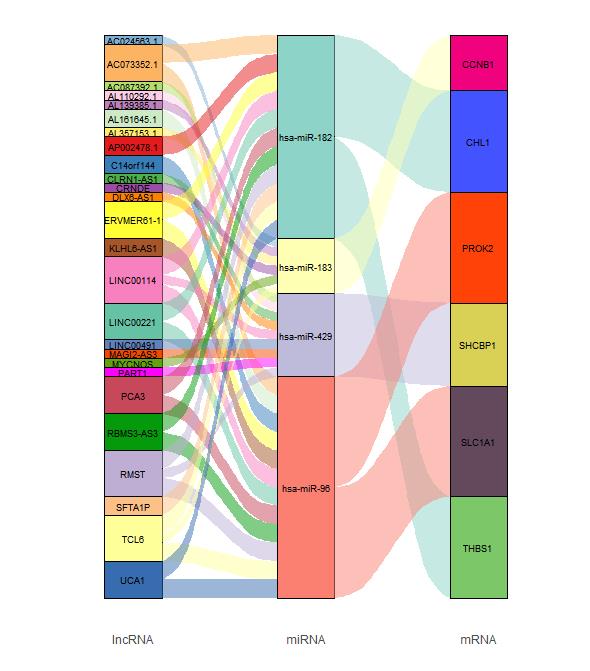
这样,文献中描述lncRNA-miRNA-mRNA关系的ceRNA冲击图就重现出来了,分子间的靶向流结构清晰,样式是不是也很漂亮呢?
此外,若老师或同学们有RNAseq(mRNA、lncRNA、miRNA、circRNA)或蛋白质组等数据分析、绘图等问题疑问,欢迎扫描下方二维码回复,我们会根据大家的需求,选择合适的问题,整理教程。
上海生因生物有着丰富的转录组测序、外显子测序数据分析的经验,同时还提供文献或分析思路整理、GEO、TCGA公共数据挖掘、高级个性化定制分析等服务。有这方面试验或数据分析需要的老师,可以添加技术微信联系我们,共同探讨如何寻找基因、分子研究,如何确定分子机制。对于已经在我们公司做过测序的老师,或者打算即将在我们公司做测序的老师,可以享受免费的售后分析服务。
关注技术微信联系数据分析

李纪伟丨写
李纪伟丨审
其他相关资料
点击阅读原文查看更多信息
以上是关于【circRNA】circRNA的鉴定的主要内容,如果未能解决你的问题,请参考以下文章
Circular RNA的产生机制功能及RNA-seq数据鉴定方法