tophat-fusion 鉴定融合基因
Posted 庐州月光
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了tophat-fusion 鉴定融合基因相关的知识,希望对你有一定的参考价值。
tophat-fusion 是一款利用RNA_seq 数据鉴定融合基因的工具,官网链接如下:
http://ccb.jhu.edu/software/tophat/fusion_index.shtml
安装:
tophat-fusion 是集成在tophat软件中的,具体的安装参考tophat的安装就好了
使用方法:
对于tophat-fusion 而言,要求固定的目录结构,比如我在result 文件夹下进行tophat-fusion的分析
那么我需要在该目录下准备几个文件:
1)物种对应的refGene.txt 和 ensGene.txt (这两个文件可以从UCSC下载得到)
2) 新建一个blast 文件夹,注意文件夹的名字必须为"blast", 在blast 文件夹下需要从NCBI下载 nt. human_genomic. other_genomic 开头的所有文件
下载的链接如下:
3) tophat_fusion 的输出目录: 每个样本一个输出目录,输出目录的前缀为tophat_, 下划线之后加上样本名称,类似 tophat_MCF7;
当然你还需要物种对应的bowtie1 的索引文件,注意这里必须为bowtie1的索引,tophat 检测融合基因时推荐bowtie1的索引方式
上述文件都准备好之后,就可以开始分析了;
第一步:toohat 比对,和普通的比对类似,只不过为了融合基因的检测,需要添加几个额外的参数:
tophat2 -o tophat_MCF7 -p 20 --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search -r 0 --mate-std-dev 80 --max-intron-length 100000 --fusion-min-dist 100000 --fusion-anchor-length 13 --fusion-ignore-chromosomes chrM hg19_bowtie1/hg19 SRR064286_1.fastq SRR064286_2.fastq
第二步:tophat-fusion-post , 生成融合基因的结果
tophat-fusion-post -p 20 --num-fusion-reads 1 --num-fusion-pairs 2 --num-fusion-both 5 hg19_bowtie1/hg19
需要指出的是,tophat-fusion-post 根据固定的目录结构进行样本,如果有多个样本,每个样本单独进行tophat 比对,只要输出目录区分开即可,比如A,B,C 3个样本,就有3个输出文件夹
tophat_A, tophat_B, tophat_C
运行完成之后,会生成一个名为 tophatfusion_out 的文件夹,该文件夹下是所有样本的融合基因分析的结果:
1)result.hml : 所有样本的融合基因分析的结果,直接看这个html
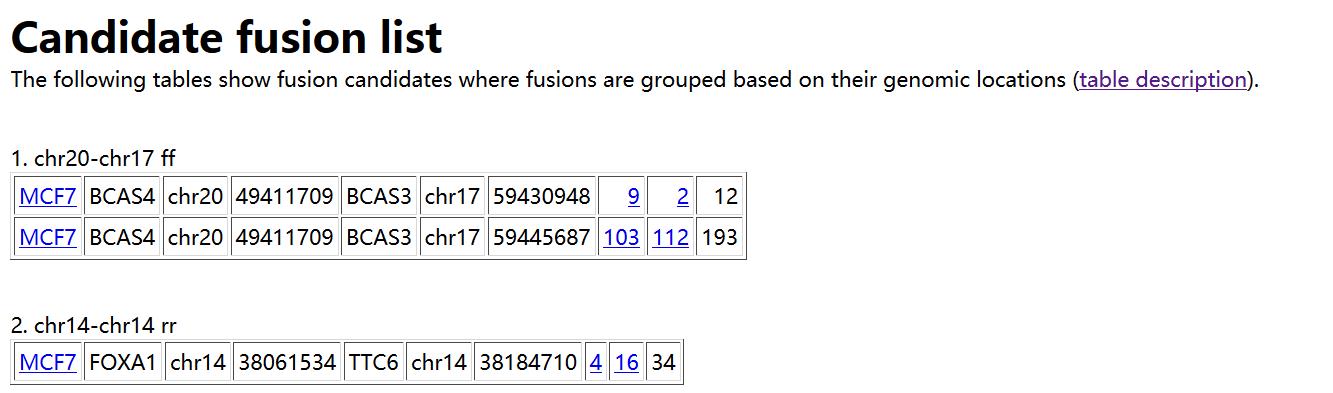
如上所示。在result.html 中,首先给出预测得到的融合基因,以表格形式进行展示,每列的含义如下:
1. Sample name in which a fusion is identified
2. Gene on the "left" side of the fusion
3. Chromosome ID on the left
4. Coordinates on the left
5. Gene on the "right" side
6. Chromosome ID on the right
7. Coordinates on the right
8. Number of spanning reads - this is the number of reads that span a
fusion point all on their own. In other words, the read itself has a
fusion break point within it.
9. Number of spanning mate pairs - this is the number of pairs of reads
where one read maps entirely on the left and the other read maps
entirely on the right of the fusion break point. Neither read is split,
so these pairs are not counted at all in (8).
10. Number of spanning mate pairs where one end spans a fusion (reads spanning fusion with only a few bases are included).
If you follow the the 9th column, it shows coordinates "number1:number2"
where one end is located at a distance of "number1" bases from the left
genomic coordinate of a fusion and "number2" is similarly defined.
以上是关于tophat-fusion 鉴定融合基因的主要内容,如果未能解决你的问题,请参考以下文章