BioNano数据的管道
Posted RandomRand
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了BioNano数据的管道相关的知识,希望对你有一定的参考价值。
BioNano数据的管道

K-INBRE生物信息学核心为BioNano分子图或预组装的BioNano基因组图谱创建了易于使用的管道,用于多次常规的组装和/或对准实验。
所有管道都有样本数据集和教程。管道从您从测绘设施收到的原始数据或汇编的基因组图谱中完成分析。
在使用这些脚本之前,不需要命令行的体验。
生物纳米分子的“原始数据到完成装配和组装分析”管线与基于序列的基因组FASTA映射
组装XeonPhi管道预处理原始分子图,并为其编写并运行一系列组件。然后用户选择最佳组装,并将其用于超级支架参考FASTA基因组文件,并总结最终装配度量和对齐。
基本步骤是首先从单个目录合并多个BNX,并绘制单分子图质量度量。然后如果参考可用,则重新缩放单分子图并绘制每次扫描的重定标因子。重新缩放步骤类似于前面的“通过扫描步骤调整拉伸扫描”。接下来,它为具有一系列参数的程序集编写脚本。组装结束后,生成装配指标,并分析最佳结果。
注意:此管道使用与AssembleIrys.pl和AssembleIrysCluster.pl相同的基本工作流程,但它运行的是具有576个内核(48x12核心Intel Xeon CPU),256GB RAM和Linux CentOS 7操作系统的Xeon Phi服务器。Assembler在不同的机器上运行BioNano可能需要进行定制。具体来说,可能需要自定义Irys-scaffolding / KSU_bioinfo_lab / assemble_XeonPhi / rescale_stretch.pl的“自定义RefAligner设置”部分,以在不同的机器上运行BioNano Assembler。也可能需要定制Irys脚手架/ KSU_bioinfo_lab / assemble_XeonPhi / clusterArguments.xml,以便程序集在不同的群集上成功运行。
请参阅教程实验室,以运行带有示例数据的组合XeonPhi管道https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/assemble_XeonPhi_LAB.md。
“BioNano分子图”的“原始数据到完成的从头装配和装配分析”管道
组装XeonPhi de novo管道预制生分子图,并为其编写并运行一系列组装。然后,用户选择最佳组件,然后总结最终组装度量。
基本步骤是首先从单个目录合并多个BNX,并绘制单分子图质量度量。接下来,它为具有一系列参数的程序集编写脚本。组装结束后,生成装配指标,并分析最佳结果。
该管道使用与AssembleIrys.pl和AssembleIrysCluster.pl相同的基本工作流程,但它运行的是具有576个内核(48x12核心Intel Xeon CPU),256GB RAM和Linux CentOS 7操作系统的Xeon Phi服务器。Assembler在不同的机器上运行BioNano可能需要进行定制。
请参阅教程实验室,以运行具有示例数据的组装XeonPhi管道https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/assemble_XeonPhi_de_novo_LAB.md。
#为BioNano数据组装提供主要的脚本
AssembleIrysXeonPhi.pl - 组装XeonPhi脚本制作原始分子映射并为其编写并运行一系列程序集。然后用户选择最佳组装,并将其用于超级支架参考FASTA基因组文件,并总结最终装配度量和对齐。
基本步骤是首先从单个目录合并多个BNX,并绘制单分子图质量度量。然后如果参考可用,则重新缩放单分子图并绘制每次扫描的重定标因子。重新缩放步骤类似于前面的“通过扫描步骤调整拉伸扫描”。接下来,它为具有一系列参数的程序集编写脚本。组装完成后,组装指标得到了分类,并对最佳结果进行了分析。
注意:此管道使用与AssembleIrys.pl和AssembleIrysCluster.pl相同的基本工作流程,但它运行的是具有576个内核(48x12核心Intel Xeon CPU),256GB RAM和Linux CentOS 7操作系统的Xeon Phi服务器。Assembler在不同的机器上运行BioNano可能需要进行定制。具体来说,可能需要自定义Irys-scaffolding / KSU_bioinfo_lab / assemble_XeonPhi / rescale_stretch.pl的“自定义RefAligner设置”部分,以在不同的机器上运行BioNano Assembler。也可能需要定制Irys脚手架/ KSU_bioinfo_lab / assemble_XeonPhi / clusterArguments.xml,以便程序集在不同的群集上成功运行。
请参阅教程实验室,以运行带有示例数据的组合XeonPhi管道https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/assemble_XeonPhi_LAB.md。
对于从头项目,请参阅本教程实验室,运行带有示例数据的组装XeonPhi管道 https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/assemble_XeonPhi_de_novo_LAB.md
新功能(相对于AssembleIrys.pl和AssembleIrysCluster.pl):
自动调整optArguments文件和迭代次数以匹配基因组大小
绘制BNX指标
减少论据数量
包括具有默认噪声参数的装配
包括新的图形,并减少装配QC文件中的细节,使选择最好的装配更容易阅读(assembly_qcXeonPhi.pl)
在可能的情况下自动组织研究人员的数据
工作流图 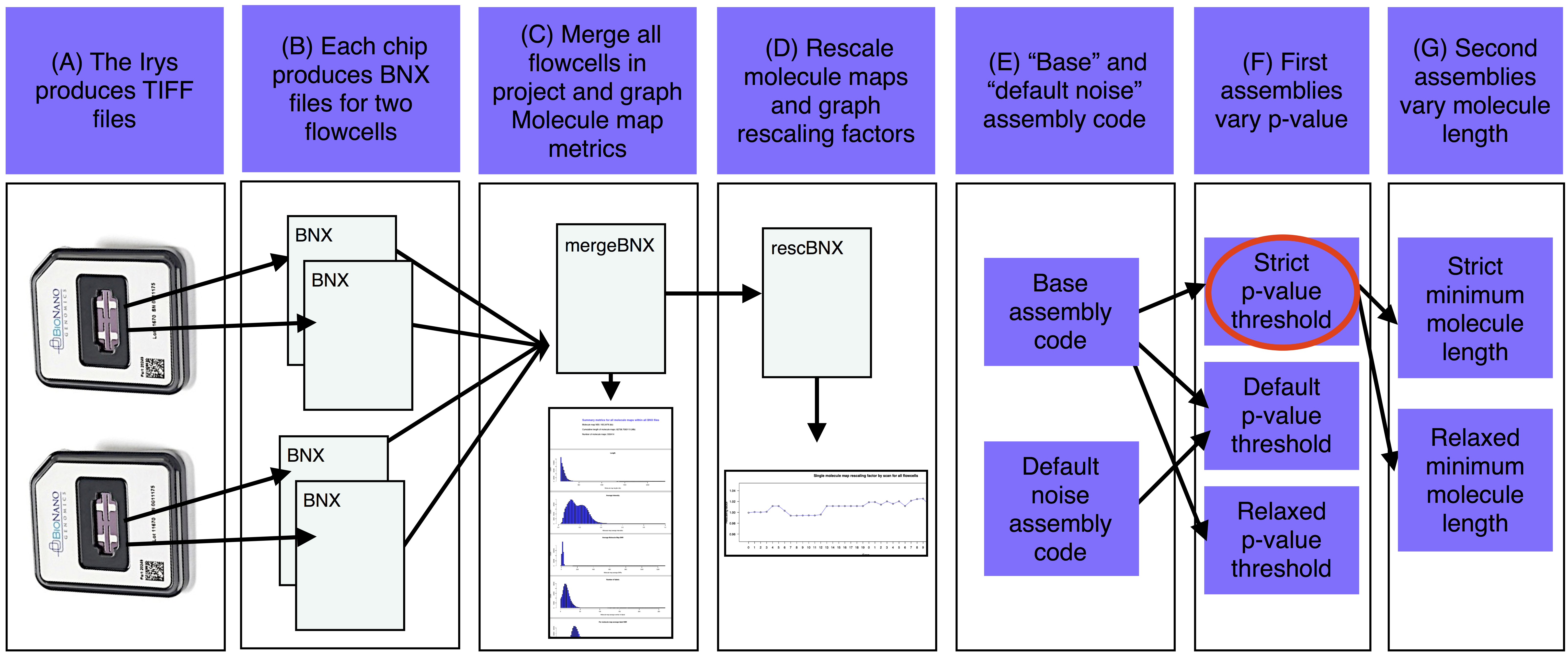
A)Irys生成的TIFF文件被转换成分子图的BNX文本文件。
B)每个IrysChip为两个流通池中的每一个生成一个BNX文件。
C)汇编工作目录bnx/子目录中的每个BNX文件-a被合并,并且分析图谱质量度量。
D)如果提供了引用,则合并的BNX文件将与序列引用中的计算机映射对齐。拉伸从对准重新缩放,并且每次扫描都会打印重新缩放系数。重新缩放的分子图与参考对齐,估计噪声参数。E)基于估计的基因组大小和噪声参数确定基本汇编代码。
F)第一个组件以各种p值阈值运行(至少有一个组件也运行有抑制噪声参数)。
G)选择最好的第一个组件(红色椭圆形),并使用各种最小分子长度过滤器生成该组件的版本。
典型用途
perl AssembleIrysXeonPhi.pl -g [genome size in Mb] -r [reference CMAP] -a [the assembly working directory for a project] -p [project name]
Usage:
perl AssembleIrysXeonPhi.pl [options]
Documentation options:
-help brief help message
-man full documentation
Required options:
-a the assembly working directory for a project
-g genome size in Mb
-r reference CMAP
-p project name for all assemblies
Optional options:
-d add this flag if the project is de novo (has no refernce)
Options:
-help Print a brief help message and exits.
-man Prints the more detailed manual page with output details and
exits.
-a, --assembly_dir
The assembly working directory for a project. This should
include the subdirectory "bnx" (any BNX in this directory will
be used in assembly). Use absolute not relative paths. Do not
use a trailing "/" for this directory.
-g, --genome
The estimated size of the genome in Mb.
-r, --ref
The full path to the reference genome CMAP.
-p, --project
The project id. This will be used to name all assemblies
-d, --de_novo
Add this flag to the command if a project is de novo (i.e. has
no reference). Any step that requires a reference will then be
skipped.以上是关于BioNano数据的管道的主要内容,如果未能解决你的问题,请参考以下文章