扩增子分析解读4去嵌合体 非细菌序列 生成代表性序列和OTU表
Posted 弗雷塞斯
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了扩增子分析解读4去嵌合体 非细菌序列 生成代表性序列和OTU表相关的知识,希望对你有一定的参考价值。
本节课程,需要先完成
先看一下扩增子分析的整体流程,从下向上逐层分析
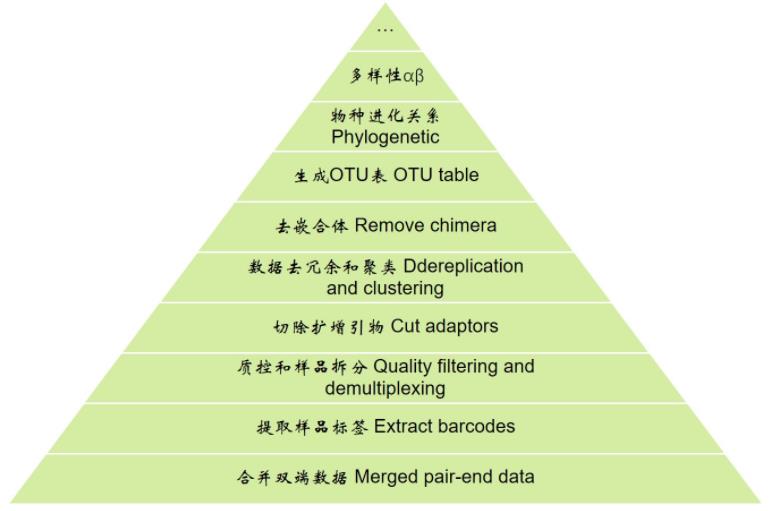
分析前准备
# 进入工作目录 cd example_PE250
上一节回顾:我们制作了Usearch要求格式的Fasta文件,对所有序列进行去冗余和低丰度过滤,并聚类生成了OTU。
接下来我们对OTU进一步去除嵌合体,并生成代表性序列和OTU表。
什么是chimeras(嵌合体)?
嵌合体序列由来自两条或者多条模板链的序列组成,示意图如下:
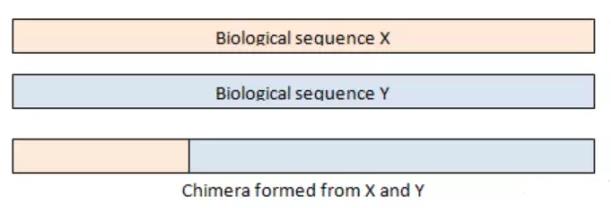
在PCR反应中,延伸阶段由于不完全延伸,就会导致嵌合体序列的出现,以上图为例,在扩增序列X的过程中,在序列延伸阶段,只产生了部分X序列延伸阶段就结束了,在下一轮的PCR反应中,这部分序列作为序列Y的引物接着延伸,扩增就会形成X和Y的嵌合体序列;
在放一张具体一点的示意图,不完全延伸产生的序列作为下一轮PCR反应的产物,进行延伸
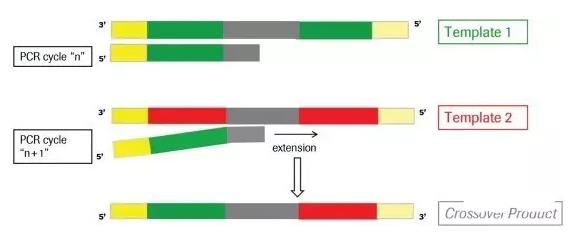
通常在PCR过程中,大概有1%的几率会出现嵌合体序列,在16S/18S/ITS 扩增子测序的分析中,系统相似度极高,嵌合体可达1%-20%,需要去除嵌合体序列。
嵌合体的比例与PCR循环数相关,循环数越高,嵌合体比例越高。
有玩过魔兽有小伙伴记得精灵族的终极兵种双头龙奇美拉吗?它的英文就是chimera,即中文的嵌合体,奇美拉是音译。
10. 基于数据库去嵌合体(可选)
上文第9步,聚OTU时,已经按照组内的序列相似情况,直接denovo去除了大量嵌合体。目前这步基于数据库去嵌合体,在以前的分析中是必做的,但随着技术发展,发现这步可能也会造成假阴性。读者可以实验设计、初步结果和预期来判断是否需要这步处理。本文示例对每一步均进行操作,即是个人风格,又是为了给大家展现一个比较全面的流程。之前Usearch作者推荐使用RDP数据去嵌合,并提供了下载链接;现在作者建议,如果做,就用Sliva或Unite这种全面的大数据库,不推荐用RDP这种小数据库,以前的建议是错的。软件方法均是不断进步的,我还没有系统比较作者的新建议有多大改进,这里还是按照原来的方法进行,读者可以自行尝试新方法。
# 下载Usearch推荐的参考数据库RDP wget http://drive5.com/uchime/rdp_gold.fa # 基于RDP数据库比对去除已知序列的嵌合体 ./usearch10 -uchime2_ref temp/otus.fa \\ -db rdp_gold.fa \\ -chimeras temp/otus_chimeras.fa \\ -notmatched temp/otus_rdp.fa \\ -uchimeout temp/otus_rdp.uchime \\ -strand plus -mode sensitive -threads 96
采用-uchime2_ref参数去嵌合体,后面接OTU序列(输入文件);
-db 指定参考数据库,这里用RDP;
-chimeras 输出检测为嵌合体的序列;
-notmatched 输出不匹配数据库的结果,即非嵌合,非相同序列;
-uchimeout 输入嵌合体的检测详细信息,如每个嵌合体的来源,与那几个亲本相似等;
-strand 指定链方向,一般为正;
-mode 选择模式,敏感的代价是嵌合体鉴定的高假阳性率;
-threads 设计线程数,程序默认系统小于10个线程为单线程;多于10个线程为10线程,根据实际情况设置,不清楚不用更好。
上面计算结果Chimeras 2669/5489 (48.6%), in db 51 (0.9%), not matched 2769 (50.4%),即5489个OTU有2669检测为嵌合、51个与数据库序列一致为非嵌合,另外2769与数据库不匹配不确定是否为嵌合。对应temp/otus_rdp.uchime文件中第三列的Y/N/?
我们想要是的排除嵌合的部分,即51+2769=2820。思路是将全部OTU中鉴定为嵌合的排除掉。
# 获得嵌合体的序列ID grep \'>\' temp/otus_chimeras.fa | sed \'s/>//g\' > temp/otus_chimeras.id # 剔除嵌合体的序列 filter_fasta.py -f temp/otus.fa -o temp/otus_non_chimera.fa -s temp/otus_chimeras.id -n # 检查是否为预期的序列数量2820 grep \'>\' -c temp/otus_non_chimera.fa
11. 去除非细菌序列(可选)
此步也是非必须的,容易造成假阴性。分析中有很多个人习惯的因素在里面,所以不同人的分析结果,也会略有不同。也缺少系统的评估到底那些更好,因为好与不好是有条件的,如何判断也不容易説清楚,这就是经验;项目经验是经过大量的项目反复研究积累出来的。
个人习惯在大数据面前,结果再多也没用,得找到有意义的东西,所以原则上是能舍即舍,更容易发现规律。万一没有发现,再回去把扔掉的捡回来试试。如果什么都不仍,规律可能永远藏在大数据的海洋中。
这步的原理是将OTU与Greengene (http://greengenes.secondgenome.com)的Align数据库比对,筛选序列相似性大于75%以上的序列作为细菌序列;此步可以排除外源非细菌的污染,非细菌序列在接下来的分析中无法注释物种分类,也很难分析。
# 下载Greengene最新数据库,320MB wget -c ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz # 解压数据包后大小3.4G tar xvzf gg_13_8_otus.tar.gz # 将OTU与97%相似聚类的代表性序列多序列比对,大约8min time align_seqs.py -i temp/otus_non_chimera.fa -t gg_13_8_otus/rep_set_aligned/97_otus.fasta -o temp/aligned/ # 无法比对细菌的数量 grep -c \'>\' temp/aligned/otus_non_chimera_failures.fasta # 1860 # 获得不像细菌的OTU ID grep \'>\' temp/aligned/otus_non_chimera_failures.fasta|cut -f 1 -d \' \'|sed \'s/>//g\' > temp/aligned/otus_non_chimera_failures.id # 过滤非细菌序列 filter_fasta.py -f temp/otus_non_chimera.fa -o temp/otus_rdp_align.fa -s temp/aligned/otus_non_chimera_failures.id -n # 看我们现在还有多少OTU:975 grep \'>\' -c temp/otus_rdp_align.fa
经过这一步过滤,从2820非嵌合的OTU,只剩下975个与细菌相似的OTU,这种数量才更接近真相。有些研究经常搞几千、几万的OTU,假阳性结果90%以上,你觉得意义何在,如何指导下游实验。
对于真菌ITS/18S,一般不建议用Unite数据库去嵌合,因为ITS/18S在所有真核生物中都有,有待物种注释后进一步确认。
12. 产生代表性序列和OTU表
代表性序列(representative sequences)即为确定的最终版的OTU,类似于参考基因组/cDNA将为索引的字典。然后将所有数据mapping于OTU上来确定各物种的丰度。
OTU表,是每个OTU在每样品中的丰度值,本质上每种高通量测序结果,都会有一个类似的表,如RNA-Seq是基因表达与样品的表
# 重命名OTU,这就是最终版的代表性序列,即Reference(可选,个人习惯)
awk \'BEGIN {n=1}; />/ {print ">OTU_" n; n++} !/>/ {print}\' temp/otus_rdp_align.fa > result/rep_seqs.fa
# 生成OTU表
./usearch10 -usearch_global temp/seqs_usearch.fa -db result/rep_seqs.fa -otutabout temp/otu_table.txt -biomout temp/otu_table.biom -strand plus -id 0.97 -threads 10
# 结果信息 01:20 141Mb 100.0% Searching seqs_usearch.fa, 32.3% matched
# 默认10线程,用时1分20秒,有32.3%的序列匹配到OTU上;用30线程反而用时3分04秒,不是线程越多越快,分发任务也是很费时间的
现在我们获得了OTU表,用less temp/otu_table.txt查看一下吧。同时还有biom可处理的标准json格式文件,用于后续分析
以上是关于扩增子分析解读4去嵌合体 非细菌序列 生成代表性序列和OTU表的主要内容,如果未能解决你的问题,请参考以下文章