Heatmap——热图那些事
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了Heatmap——热图那些事相关的知识,希望对你有一定的参考价值。
参考技术A 注脚:
scale 标准化,根据每列或者行数据的均值标准化,主要是为了防止单个数据过大(过小),导致冷热色分布不明显的现象,可选row,column,none。
**key 是否需要图标以及图标大小,key=T或者F,keysize=1.5;
Colv=NA 表示不对行聚类,Rowv=NA不对列聚类
dendrogram =‘column’or \'none\'or\'row\' or"both"显示聚类树状图,
trace ,是否需要基准线(均值,方差之类的),trace="both","row" 或者"column"
density.info 指示图内的线, density.info=\'none\'
cexCol=1,cexRow=1 设置xlab和ylab的字符大小
#col ,优化颜色:redgreen或者greenred, 调整配色,括号内表示这个配色区间分成多少格区分度; breaks=seq 自己设置颜色分度breaks=seq(-5, 5, 1))
** labels,labCol调整每列的标记 labCol = NA,或者 labCol = c(\'Age\', \'A\', \'B\', \'C\'))
margins 调整画布边距; margins=c(5,5),
main =\'Heatmap\'增加标题。
注脚:
第一个参数是需要用pheatmap画图的数据
color: 设置颜色。精细程度按照括号内设置的数值来定
main: 标题名称
fontsize: 设置row的字体大小
scale: 设置归一化为正态分布,可选row,column,none。
border_color: 是否显示边框及边框的颜色,NA不显示,red显示红色。支持简单的颜色单词
na_col: 设置缺失值的颜色,支持简单颜色单词,一般设置为灰色就满好识别的。
cluster_rows&cluster_cols: 设置是否对行或者列进行聚类,按照实际需求设置。当缺失值较多的时候是无法进行聚类的。**一个解决办法是读取数据的时候不设置缺失值。
show_rownames&show_colnames: 是否显示行/列的名称
treeheight_row&treeheight_col: 当前面设置了聚类之后,两边会出现聚类的树,这个参数是设置树的高度的。
cellheight&cellwidth: 设置每个各自格的宽度和高度。
cutree_row&cutree_col: 是否根据聚类情况把树切开,可以设置切开的份数。
display_numbers: 设置是否显示每个单元格的值。
legend: 设置是否显示旁边的bar状图例。
filename: 设置输出文件的名字。可以设置的文件类型有:pdf,png,jpg,tiff,bmp。
优点:1、支持多种的颜色配置;2、支持样品和基因的双聚类;
缺点:基因名称显示似乎不太行,还在尝试,数据上限:2,500 rows and 300 columns;
操作参考-没有,慢慢摸索就好,简单易上手,似乎没有数据上限?
似乎全英,看着好累
这个工具看着也蛮好用的,操作参考也很详细,全中文参考: 重磅推荐,超详细热图在线绘制教程资料! - ,不过我不想注册,就没尝试
用R包中heatmap画热图
一:导入R包及需要画热图的数据
library(pheatmap)
data<- read.table("F:/R练习/R测试数据/heatmapdata.txt",head = T,row.names=1,sep=" ")
二:画图
1)pheatmap(data)#默认参数
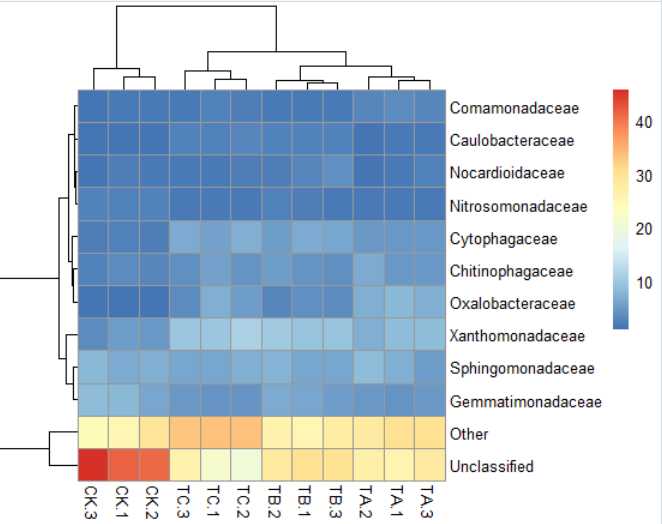
2)pheatmap(data,clustering_distance_rows = "correlation")#聚类线长度优化
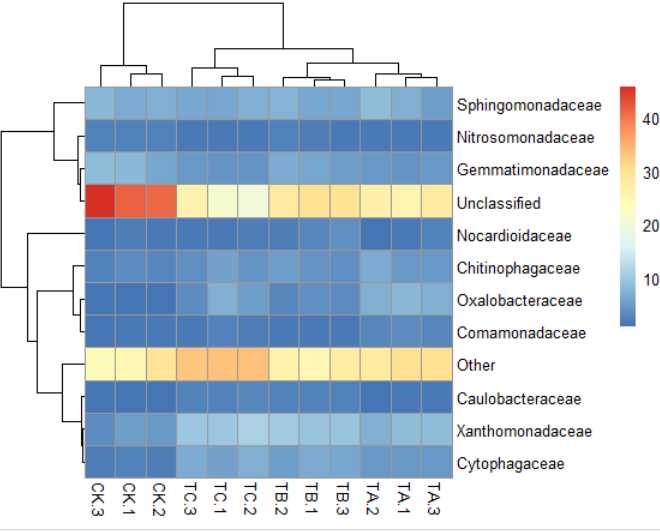
3)pheatmap(data,scale="column")#按列均一化,"row","column" or "none"默认是"none"
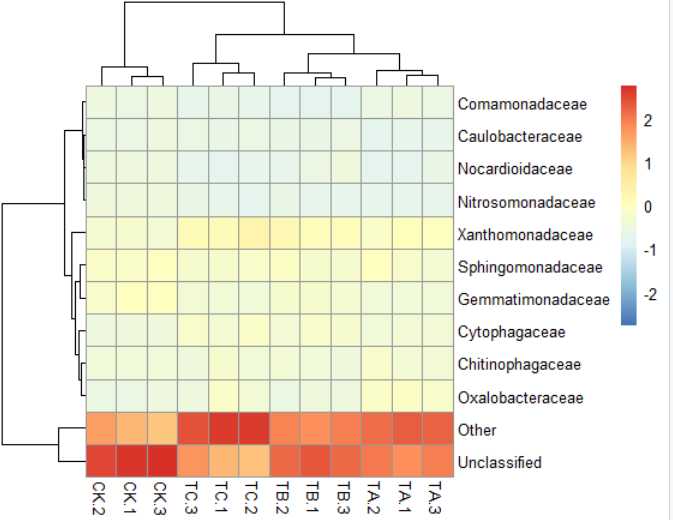
4)pheatmap(data,scale="row")#按行均一化,"row","column" or "none"默认是"none"
5)pheatmap(data,display_numbers=T,number_format="%.2f",number_color="red",fontsize_number=8)#是否在每一格上显示数据,及其数据格式,大小及其颜色
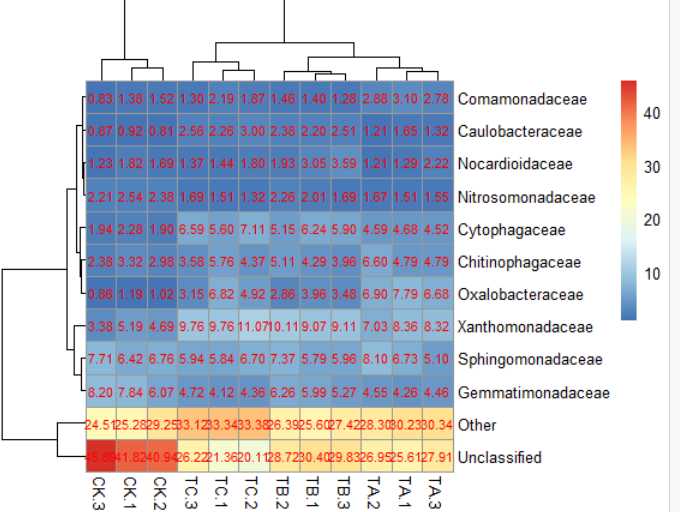
6)pheatmap(data,cellwidth = 50,cellheight= 14)#格子大小
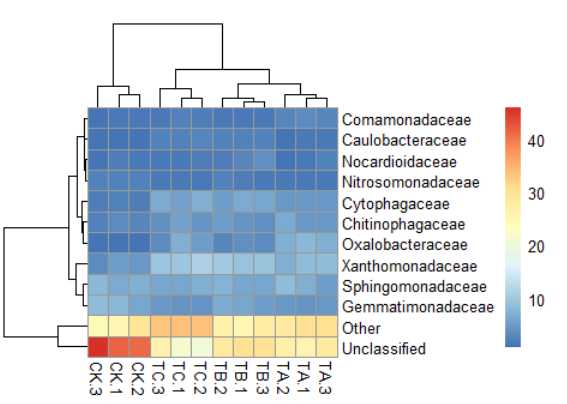
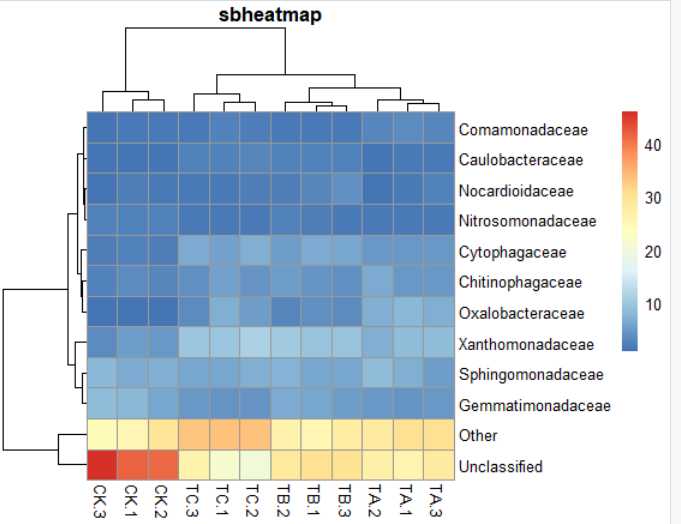
7) pheatmap(data,main="sbheatmap")#标题
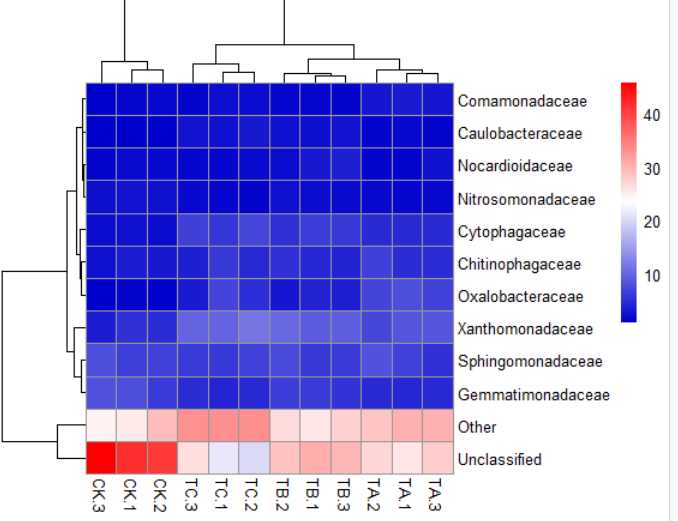
8)pheatmap(data,color = colorRampPalette(c("MediumBlue","white","red"))(256))#颜色
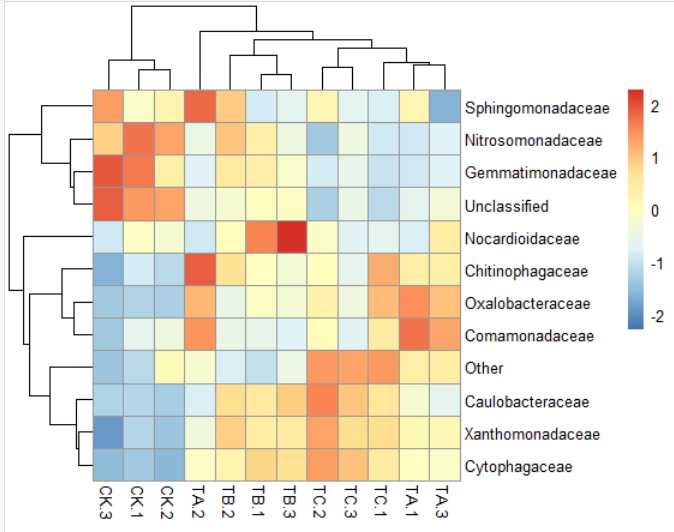
9)pheatmap(data,clustering_distance_rows = "correlation",scale="column",display_numbers=T,number_format="%.1f",number_color="black",
fontsize_number=8,cellwidth = 14,cellheight= 14,color = colorRampPalette(c("MediumBlue","white","red"))(256))

图3 略丑陋的双列热图
原因是当每行只有两个数值的时候,任何两个不同的数值标准正态均一化后,都会变成-1和1。所以,会产生上述的图形(只有两种颜色)。面对这种类型的数据,建议直接计算每行两个数值的倍数的log2值,然后使用OS-tools画单列的热图。
当然, 单列热图也可以使用R语言的pheatmap包绘制,并通过一个函数控制0点的位置,在另一个R语言绘图的主题帖中也有介绍。
(3)聚类的原理
以上是关于Heatmap——热图那些事的主要内容,如果未能解决你的问题,请参考以下文章