单细胞36计之5趁火打劫---锚点整合
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了单细胞36计之5趁火打劫---锚点整合相关的知识,希望对你有一定的参考价值。
参考技术A 对两个或多个单细胞数据集的联合分析提出了独特的挑战。特别是,在标准工作流程下,识别多个数据集中存在的细胞群体可能会成问题。Seurat v4包括一组用于匹配(或“对齐”)跨数据集的共享细胞群体的方法。这些方法首先确定处于匹配生物学状态(“锚”)的细胞的跨数据集对,既可以用于校正数据集之间的技术差异(即批效应校正),也可以用于对基因组进行比较性scRNA-seq分析跨实验条件。下面,我们展示了 Stuart *,Butler *等人,2019中 所述的scRNA-seq整合方法,以对处于 静止或干扰素刺激状态 的人免疫细胞(PBMC)进行比较分析。
以下教程旨在概述使用Seurat集成过程可能进行的复杂细胞类型的比较分析。在这里,我们解决了一些关键目标:
为了方便起见,我们通过 SeuratData 软件包分发此数据集。
然后,我们使用 FindIntegrationAnchors() 函数来识别锚点,该函数将Seurat对象的列表作为输入,并使用这些锚点将两个数据集与集成在一起 IntegrateData() `。
现在,我们可以在所有单元上运行单个集成分析!
为了并排可视化这两个条件,我们可以使用 split.by 参数来显示每个以聚类着色的条件。
为了鉴定在各种条件下保守的规范细胞类型标记基因,我们提供了该 FindConservedMarkers() `功能。此功能对每个数据集/组执行差异基因表达测试,并使用MetaDE R软件包中的荟萃分析方法组合p值。例如,无论簇6中的刺激条件如何,我们都可以计算出保守标记的基因(NK细胞)。
我们可以为每个簇探索这些标记基因,并使用它们将我们的簇注释为特定的细胞类型。
DotPlot() 带有 split.by`参数的函数可用于查看各种条件下的保守细胞类型标记,显示表达水平和表达任何给定基因的簇中细胞的百分比。在这里,我们为14个簇中的每个簇绘制2-3个强标记基因。
现在,我们已经排列了刺激细胞和对照细胞,我们可以开始进行比较分析,并观察刺激引起的差异。广泛观察这些变化的一种方法是绘制受刺激细胞和对照细胞的平均表达,并在散点图上寻找视觉异常值的基因。在这里,我们采用受刺激的和对照的原始T细胞和CD14单核细胞群体的平均表达,并生成散点图,突出显示对干扰素刺激表现出戏剧性反应的基因。
如您所见,许多相同的基因在这两种细胞类型中均被上调,可能代表保守的干扰素应答途径。
因为我们有信心确定出跨条件的常见细胞类型,所以我们可以询问相同条件下不同条件下哪些基因会发生变化。首先,我们在meta.data插槽中创建一列,以保存细胞类型和刺激信息,并将当前标识切换到该列。然后,我们用于 FindMarkers() `查找受激B细胞和对照B细胞之间不同的基因。请注意,此处显示的许多顶级基因与我们之前绘制的核心干扰素应答基因相同。此外,我们看到的像CXCL10的基因对单核细胞和B细胞干扰素的反应也具有特异性,在该列表中也显示出很高的意义。
可视化基因表达中这些变化的另一种有用方法是 split.by 选择 FeaturePlot() 或 VlnPlot() 功能。这将显示给定基因列表的FeaturePlots,并按分组变量(此处为刺激条件)进行划分。诸如CD3D和GNLY之类的基因是典型的细胞类型标记(对于T细胞和NK / CD8 T细胞),实际上不受干扰素刺激的影响,并且在对照组和受刺激组中显示出相似的基因表达模式。另一方面,IFI6和ISG15是核心干扰素反应基因,因此在所有细胞类型中均被上调。最后,CD14和CXCL10是显示细胞类型特异性干扰素应答的基因。CD14单核细胞受刺激后,CD14表达下降,这可能导致在有监督的分析框架中进行错误分类,从而强调了整合分析的价值。
[图片上传失败...(image-275df6-1615650992246)]
单细胞分析:数据整合
导读
本文将学习跨条件执行单细胞整合,以识别彼此相似的细胞。
1. 目标
-
跨条件对齐相同的细胞类型。
2. 挑战
对齐相似细胞类型的细胞,这样就不会因为样本、条件、模式或批次之间的差异而在后续分析中进行聚类。
3. 推荐
建议先不整合分析,再决定是否进行整合。
4. 整合与否
通常,在决定是否需要执行任何对齐之前,我们总是在没有整合的情况下查看聚类。不要仅仅认为可能存在差异而总是先执行整合,探索数据。如果在 Seurat 对象中同时对两种条件进行归一化并可视化细胞之间的相似性,会看到特定条件的聚类情况:
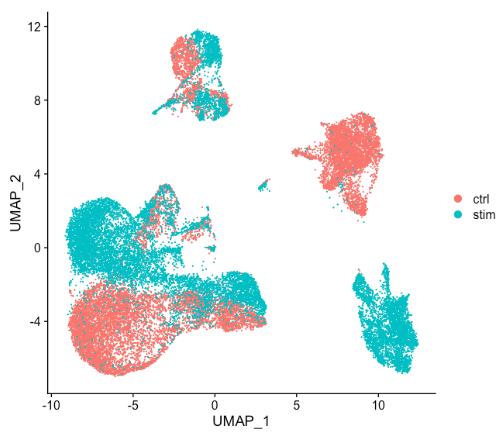
细胞在特定条件下聚类表明需要跨条件整合细胞以确保相同类型的细胞聚集在一起。
-
为什么相同细胞类型的细胞聚集在一起很重要?
想要识别存在于数据集中所有的细胞类型,因此希望观察每个簇中两个样本/条件/模态中的细胞表示。这将使下游的结果更具可解释性(即 DE 分析、配体-受体分析)。在本课中,将介绍跨条件的样本整合,该教程改编自 Seurat v3 Guided Integration Tutorial[1]。
注意:
Seurat有一个关于如何在不整合的情况下运行工作流程的小插图。工作流程与此工作流程非常相似,但样本不一定在一开始就被拆分,也不会执行整合。如果不确定在条件(例如肿瘤和对照样本)之间会出现什么簇或预期某些不同的细胞类型,则首先单独运行会有所帮助,然后将它们一起运行以查看两种条件下是否存在针对细胞类型的特定条件簇。通常,当对来自多个条件的细胞进行聚类时,会有特定于条件的聚类,而整合有助于确保相同的细胞类型聚类在一起。
5. 整合
利用共享的高可变基因跨条件整合或对齐样本。
如果细胞按样本、条件、批次、数据集、模态进行聚类,则整合步骤可以极大地改善聚类和下游分析。
为了整合,将使用来自每个组的高可变基因(使用 SCTransform 识别),然后,将“整合”或“协调”这些组以覆盖相似或具有“共同生物特征集”的细胞团体。例如,可以整合:
-
不同条件(例如对照和处理):
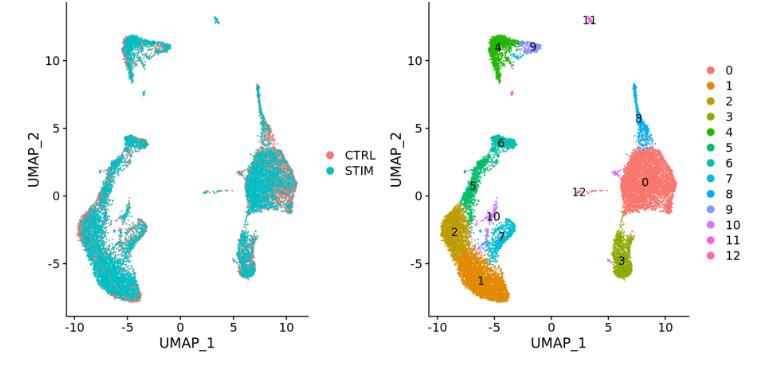
-
不同数据集(例如,来自在相同样本上使用不同文库制备方法生成的 scRNA-seq数据集):
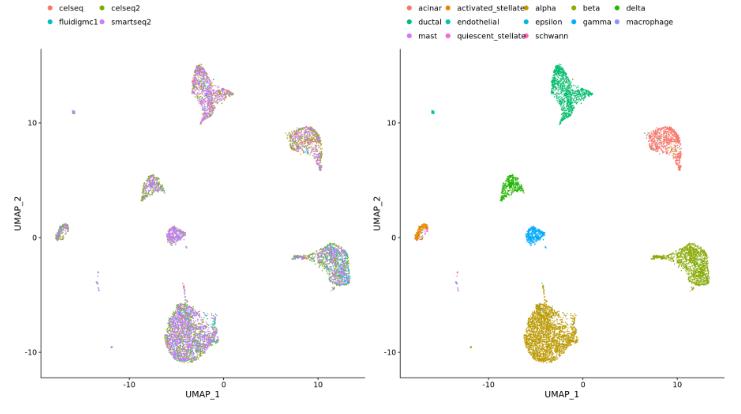
-
不同的组学数据(例如 scRNA-seq和scATAC-seq):
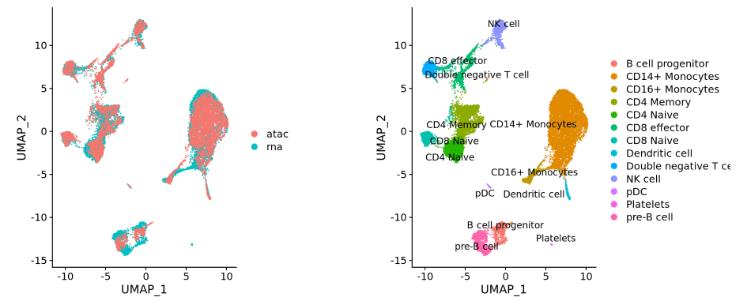
-
不同批次(例如,当实验条件需要对样品进行批量处理时)
整合是一种强大的方法,它使用这些最大变异的共享源,来识别跨条件或数据集的共享亚群。整合的目标是确保一个条件/数据集的细胞类型与其他条件/数据集的相同细胞类型对齐(例如,控制巨噬细胞与受刺激的巨噬细胞对齐)。
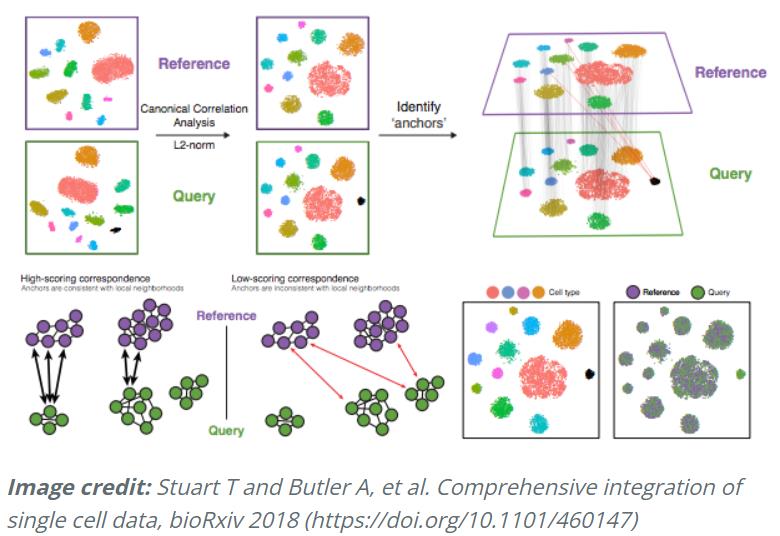
具体来说,这种整合方法期望组中至少一个单细胞子集之间存在“对应”或共享的生物状态。整合分析的步骤如下图所示:
-
应用的不同步骤如下:
-
典型相关分析 ( CCA):
CCA 识别条件/组之间的共享变异源。它是 PCA 的一种形式,因为它可以识别数据中最大的变异来源,但前提是它在条件/组之间共享或保存(使用来自每个样本的 3000 个变异最多的基因)。
此步骤使用最大的共享变异源大致对齐细胞。
注意:使用共享的高度可变基因是因为它们最有可能代表那些区分存在的不同细胞类型的基因。
-
跨数据集识别 anchors或mutual nearest neighbors(MNN)(有时会识别出不正确的anchors):
MNN 可以被认为是best buddies。对于一个条件下的每个细胞:
-
在另一种情况下,细胞最近的邻居是根据基因表达值确定的。 -
执行相互分析,**如果两个细胞在两个方向上都是 best buddies,那么这些细胞将被标记为anchors**,以将两个数据集“锚定”在一起。
“
MNN对细胞之间表达值的差异提供了对批次效应的估计,通过对许多这样的对进行平均可以更加精确。获得一个校正向量并将其应用于表达式值以执行批量校正。”
-
过滤 anchors以删除不正确的anchors:
通过本地邻域中的重叠来评估anchors对之间的相似性(不正确的anchors得分会很低)
-
整合条件/数据集:
使用anchors和相应的分数来转换细胞表达式值,允许整合条件/数据集(不同的样本、条件、数据集、模态)。
注意:每个细胞的转换使用跨数据集
anchors的每个anchors的两个细胞的加权平均值。权重由细胞相似度得分(细胞与 k 个最近anchors之间的距离)和anchors得分确定,因此同一邻域中的细胞应该具有相似的校正值。
如果细胞类型存在于一个数据集中,但不存在于另一个数据集中,则细胞仍将显示为单独的样本特定簇。
现在,使用 SCTransform 对象作为输入,执行跨条件的整合。
首先,需要指定使用 SCTransform 识别的所有 3000 个可变基因进行整合。默认情况下,此函数仅选择前 2000 个基因。
# 选择用于整合的可变性最大的特征,
integ_features <- SelectIntegrationFeatures(object.list = split_seurat,
nfeatures = 3000)
现在,需要为整合准备SCTransform对象。
# 准备 SCT 列表对象以进行整合
split_seurat <- PrepSCTIntegration(object.list = split_seurat,
anchor.features = integ_features)
现在,将执行 CCA,找到best buddies或anchors并过滤不正确的anchors。请注意控制台中的进度条将保持在 0%,但它实际上正在运行。
# 寻找`best buddies` - 可能需要一段时间才能运行完毕
integ_anchors <- FindIntegrationAnchors(object.list = split_seurat,
normalization.method = "SCT",
anchor.features = integ_features)
最后,跨条件整合。
# 跨条件整合
seurat_integrated <- IntegrateData(anchorset = integ_anchors,
normalization.method = "SCT")
6. UMAP 可视化
整合后,为了可视化整合数据,可以使用降维技术,例如 PCA 和UMAP。虽然 PCA 将确定所有 PC,但一次只能绘制两个。相比之下,UMAP 将从任意数量的顶级 PC 获取信息,以在这个多维空间中排列细胞。它将在多维空间中获取这些距离,并将它们绘制成二维,以保持局部和全局结构。这样,细胞之间的距离代表了表达的相似性。如果想更详细地探索 UMAP[2],这篇文章是对 UMAP 理论的一个很好的介绍。
生成这些可视化,首先运行 PCA 和 UMAP 方法。从 PCA 开始。
# 运行 PCA
seurat_integrated <- RunPCA(object = seurat_integrated)
# 画图
PCAPlot(seurat_integrated,
split.by = "sample")
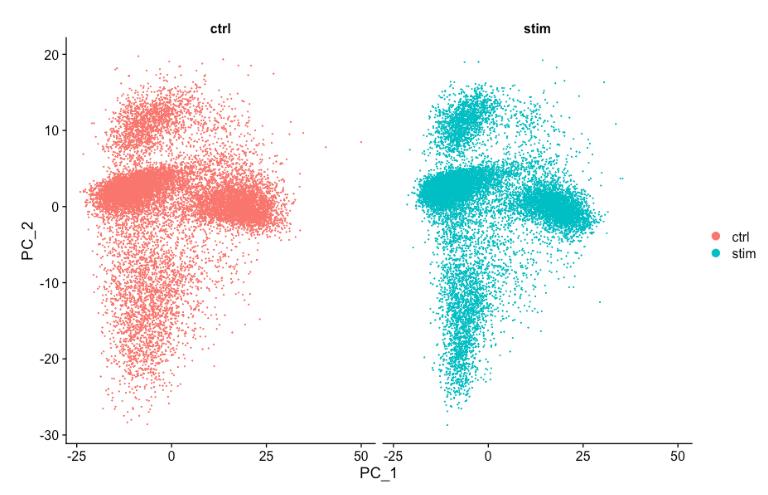
通过 PCA 映射,可以看到 PCA 很好地覆盖了这两个条件。
现在,使用 UMAP 进行可视化。
# 运行 UMAP
seurat_integrated <- RunUMAP(seurat_integrated,
dims = 1:40,
reduction = "pca")
# 画图
DimPlot(seurat_integrated)
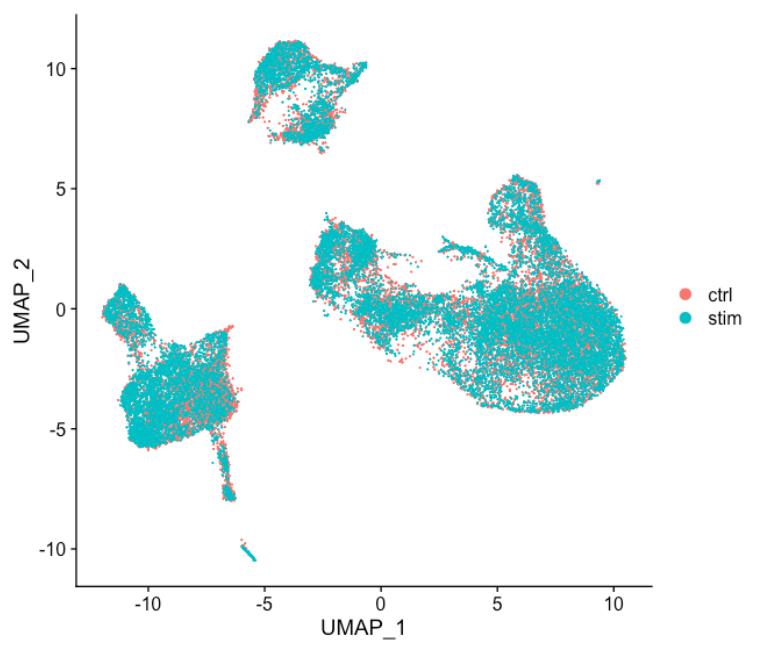
当将上图中的 ctrl 和 stim 细胞类群之间的相似性与未整合数据集(下图)进行比较时,很明显整合有益于分析!
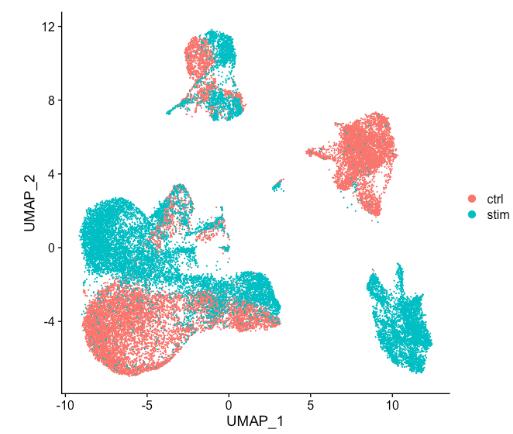
-
细胞类群的并排比较
有时,如果在条件之间拆分绘图,则更容易查看所有细胞是否对齐良好,可以通过将 split.by 参数添加到DimPlot()函数来做到这一点:
# 通过样本分割 UMAP
DimPlot(seurat_integrated,
split.by = "sample")
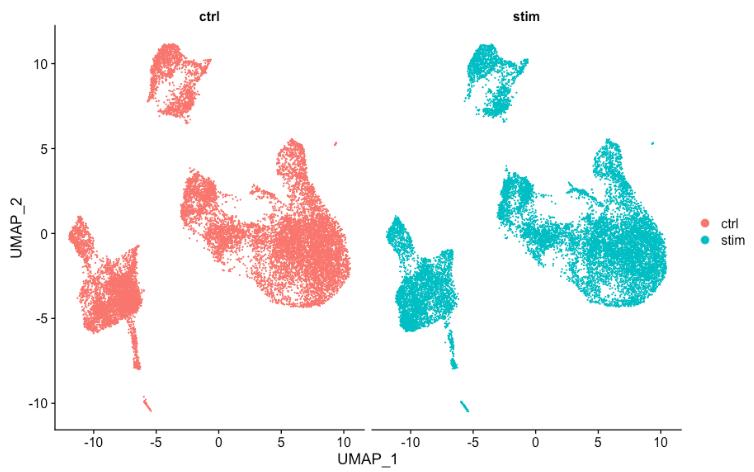
7. 保存
由于整合可能需要一段时间,因此保存整合的 seurat 对象通常是一个好主意。
saveRDS(seurat_integrated, "results/integrated_seurat.rds")
参考资料
Seurat: "https://satijalab.org/seurat/v3.0/immune_alignment.html"
[2]UMAP: https://pair-code.github.io/understanding-umap/
本文由 mdnice 多平台发布
以上是关于单细胞36计之5趁火打劫---锚点整合的主要内容,如果未能解决你的问题,请参考以下文章
10X单细胞转录组整合、转录组 && ATAC整合分析之VIPCCA