GWAS文献基于GWAS与群体进化分析挖掘大豆相关基因
Posted zkkaka
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GWAS文献基于GWAS与群体进化分析挖掘大豆相关基因相关的知识,希望对你有一定的参考价值。
发表期刊杂志:nature biotechnology
影响因子:41.514
发表时间:2015年2月
发表单位:中科院遗传与发育生物学研究所
一、 研究取材
62株野生大豆、130株地方种和110个驯化品种构建的一个自然群体
二、 方法流程
Illumina HiSeq 2000 测序平台,测序文库300bp,样本平均测序深度达到11X
三、 生物信息学分析
群体结构分析、选择清除分析、重要性状的全基因组关联分析
四、 研究结果
1)使用BWA软件将原始数据与参考基因组进行比对,使用samtools将sam格式转化为bam,使用picard软件去掉Duplicated reads。
2)SNP calling使用GATK和samtools,取两者结果的交集。对于GATK参数设置:-stand_call_conf 30。MAF设置为0.01。
3) Indel calling类似于SNP calling,使用GATK的UnifiedGenotyper程序,起参数设置为-glm INDEL,只考虑6bp范围内的缺失和插入。
4)SNP注释使用的软件为ANNOVAR。SNP被注释到内含子(overlap- ping with an intron)、外显子、基因间区,可变剪切位点(within 2 bp of a splicing junction)、5′UTRs 、3′UTRs,, upstream and downstream regions (within a 1 kb region upstream or downstream from the transcription start site).注释在外显子区域的SNP又分为同义和非同义突变。注释到外显子的Indel又分为移码突变和非移码突变。
5)群体结构分析中,PCA使用的是EIGENSOFT 4.2 的smartpca 程序,neighbor-joining tree 使用PHYLIP 3.68软件。结构分层使用FRAPPE,其中k值选取2到7.连锁不平衡分析使用plink软件。关联分析使用的GAPIT 分析软件。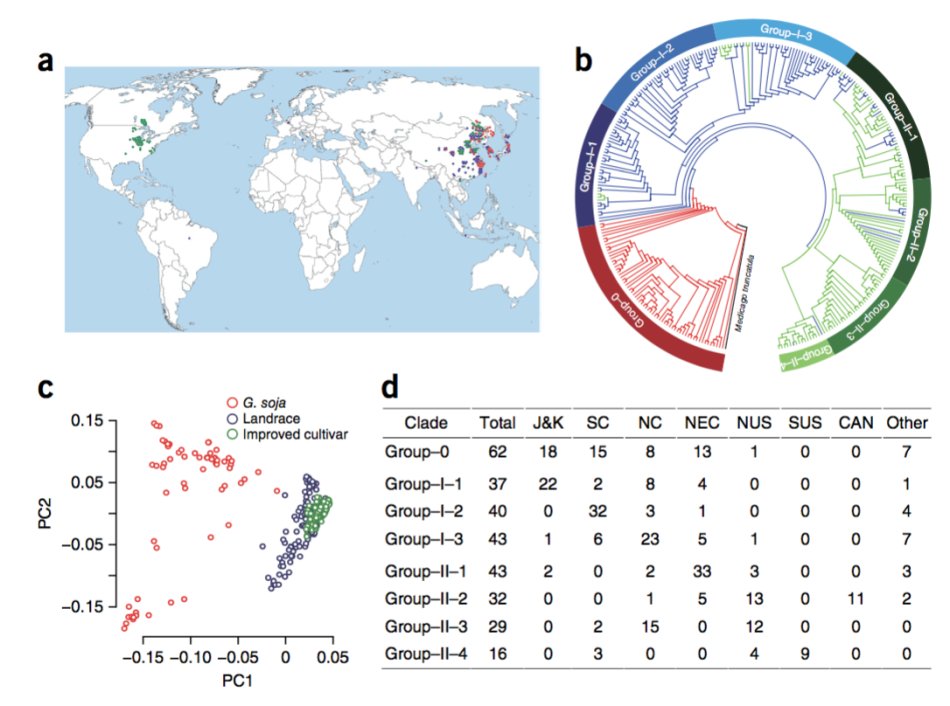
以上是关于GWAS文献基于GWAS与群体进化分析挖掘大豆相关基因的主要内容,如果未能解决你的问题,请参考以下文章