序列比对
Posted djx571
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了序列比对相关的知识,希望对你有一定的参考价值。
最常用的产生多重对齐的算法是从渐进对齐方法中派生出来的。这是Fitch和Yasunobu(1975)提出的,Hogeweg和Hesper(1984)将其应用于5S核糖体的比对RNA序列。该方法由 Da-Fei Feng and Russell Doolittle 推广。它被称为“渐进”,因为该策略需要计算所有对齐的蛋白质(或核酸序列)之间的成对序列比对得分,然后从两个最近的序列开始比对,并逐步向比对中添加更多的序列。这种方法的一个好处是允许数百甚至数千个序列快速对齐。一个主要的限制是,最终的对齐取决于序列连接的顺序。因此,它不能保证提供最精确的对齐。从20世纪90年代到最近,最流行的基于web的渐进多序列比对程序是ClustalW
一、渐进式比对(progressive Sequence alignment)
虽然大多数专家推荐更新的程序(如MAFFT,提供改进性能的ProbCons、MUSCLE和T-COFFEE),我们引入ClustalW算法来解释渐进式比对。它分三个阶段进行。
1.1、执行成对比对的算法生成原始相似性评分
注意,对于ClustalW的默认设置,分数只是百分比标识。包括ClustalW在内的许多渐进序列对齐算法都使用距离矩阵,而不是用相似矩阵来描述蛋白质的亲缘关系。框6.1中概述了每对序列的相似度分数到距离分数的转换。生成距离度量的目的是生成一个导向树(下面是第二阶段)来构建对齐。
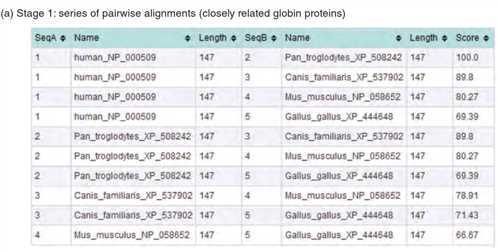
1.2、在第二阶段,从距离(或相似度)矩阵计算导树。构建指导树的主要方法有两种:算术平均的非加权对群法(UPGMA)和邻接法
在第二阶段,从距离(或相似度)矩阵计算导树。构建指导树的主要方法有两种:算术平均的非加权对群法(UPGMA)和邻接法。树的两个主要特征是它的拓扑结构(分支顺序)和分支长度(可以画出它们与进化距离成正比)。因此,这棵树反映了所有要排列的蛋白质的亲缘关系。在ClustalW中,用一种称为Newick格式的书面语法以及图形输出来描述树。指导树通常不被认为是真正的系统发育树,而是ClustalW的第三阶段中使用的模板,用于定义序列添加到多个对齐的顺序。基于序列比对的相似率转化成距离矩阵生成指导树( A guide tree is estimated from a distance matrix based on the percent identities between sequences you are aligning)。相比之下,系统发育树几乎总是包含一个模型来解释同时发生在氨基酸(或核苷酸)排列位置的多个取代(多重替代)。
1.3、在阶段3中,根据指导树中给出的顺序,通过一系列步骤创建多个序列对齐
该算法首先从指导树中选择两个最接近的序列,并创建pairwise alignment。这两个序列出现在树的终端节点,即现存序列的位置。下一个序列要么添加到pairwise alignment(生成由三个序列组成的对齐组),要么用于另一个pairwise alignment。alignment将逐步进行,直到树的根到达,并且所有序列都已对齐。此时将获得完整的多序列比对。
Feng–Doolittle的方法包括这样一条规则:“一次gap,永远是gap。最closely的一对序列首先比对。随着进一步的序列被添加到比对中,有许多方法可以包含gap。“一次gap,永远是gap”规则的基本原理是,在分配gap时,最紧密相关的两个序列(大部分对齐)的权重应该最大。ClustalW动态地分配特定于位置的gap罚分,这增加了在与现有gap相同的位置出现新gap的可能性。这就为整体对齐提供了一个类似块的结构,在将间隙位置的数量最小化方面,这种结构常常显得非常有效。
插入和删除应该受到相同的惩罚吗?不,据Loytynoja和Goldman(2005):一个单一的删除事件通常会在它发生的地方受到罚分,但是一个不恰当的插入事件一旦发生,就会对所有其他序列造成多重惩罚。这些高罚分的结果是许多序列比对不切实际地比对,with too few gaps。Loytynoja和Goldman(2005)引入了一对隐马尔可夫模型方法来区分插入和删除。他们表示,他们的方法创造了与系统发育相一致的gap,尽管这些排列看起来没有ClustalW紧凑。他们的方法适用于蛋白质、RNA或DNA序列的比对,但对基因组DNA的比对可能特别有用。在这种情况下,传统的渐进式对齐可能会发生过拟合,例如当一个序列有很长的内嵌时。Loytynoja和Goldman(2005)的方法,在Higgins等人(2005)的综述中,提供了多个序列比对,这些序列比对有更多的gap,但基于外显子的正确比对等标准,可能更准确。
ClustalW实现了一系列附加功能来优化对齐。计算每个蛋白质(或DNA)序列到指导树根的距离,并将那些最密切相关的序列按乘性因子加权。这种调整确保,如果一个比对包括一组非常密切相关的序列以及另一组发散序列,那么密切相关的序列不会过多地支配最终的多个序列比对。其他调整包括使用一系列评分矩阵,这些矩阵根据蛋白质的相似性应用于成对排列,并补偿序列长度上的差异。许多其他算法使用渐进式对齐的变体。
二、迭代式(Iterative approaches)
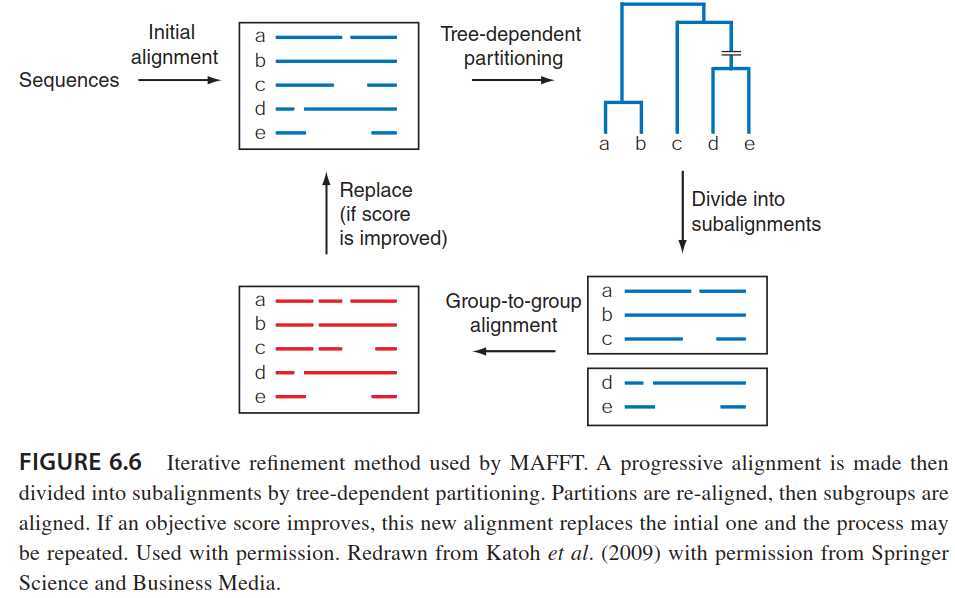
迭代方法使用渐进比对策略计算次优解,然后使用动态规划或其他方法修改比对,直到解收敛。对初始树进行划分,并重新比对每一边的profiles。因此,这些方法创建一个初始对齐,然后修改它,试图改进它,使用一些目标函数来最大化得分。渐进式比对方法有其固有的局限性,即比对过程中一旦发生误差,就无法纠正;迭代方法可以克服这种限制。在标准动态规划中,指导树的分支顺序可能不是最优的,或者评分参数可能导致gap错位。迭代求精可以随机地搜索更多的最优解(根据某些度量,如 sum-of-pairs scores 或SPS,寻找更高的最大得分),或者通过系统地从生成的初始profile文件中提取和重新组合序列。使用迭代方法的程序的例子是MAFFT, Iteralign, PRALINE,and MUSCLE 。
2.1、maffat
MAFFT是一个多对齐包的例子,根据最近的基准测试研究,它被认为是非常精确的。它提供了一套工具,可以选择更快或更准确的速度。它包括渐进式比对,包括:(1)一个单周期的渐进式方法(调用FFT-NS-1)类似ClustalW,但对细化步骤使用快速傅里叶变换。(2)一种双循环方法(FFT-NS-2),在该方法中创建多个比对,然后从该比对计算细化距离,形成第二个渐进比对;(3)一个名为PartTree的非常快速的渐进式对准器,用于对齐大量序列(∼50,000)。渐进式对齐使用匹配的6元组(六残基串)计算成对距离。这种方法称为k-mer计数。k-mer(也称为k元组或单词)是长度为k的连续子序列。k-mer计数非常快,因为它不需要比对。初始距离矩阵可以重新计算,一旦所有pairwise alignments的计算,产生一个更可靠的渐进比对。在迭代求精步骤中,一个加权的sum-of-pairs score被计算及最优化。MAFFT允许包括全局或局部比对在内的选项。
MAFFT和PRALINE都可以合并来自同源序列的信息,这些信息除了您提交的用于多个序列比对的信息之外,还可以对其进行分析。这些序列用于改进多序列比对;在MAFFT的情况下,多余的序列将被删除。PRALINE对查询的蛋白质序列执行PSI-BLAST搜索,然后使用PSI-BLAST概要文件。PRALINE还允许合并预测的二级结构信息。
罗伯特·埃德加(Robert Edgar, 2004a, 4b)的MUSCLE program自2004年推出以来,由于其准确性和超常的速度,特别是对于涉及大量序列的多个序列比对,已经变得流行起来。例如,在台式计算机上,1000个平均长度282个残基的蛋白质序列在21秒内对齐(Edgar, 2004a)。
2.2、MUSCLE
2.2.1、生成一个渐进比对。
为了实现这一点,该算法使用similarity(从每对序列的全局对齐中计算)或k-mer计数来计算每对序列之间的相似性。根据相似点,MUSCLE计算一个三角形距离矩阵,然后使用UPGMA或邻域连接构造一个有根的树。基于相似之处,MUSCLE计算一个三角形距离矩阵,然后使用UPGMA或邻域连接构造一个有根的树。序列按照树的分支顺序被添加到多个序列对齐中。
2.2.2、MUSCLE改进树,并构建一个新的渐进对齐(或一组新的对齐)
用fractional identity对每对序列的相似性进行了评价,然后用Kimura distance matrix构建一棵树。在两个序列的比较中,有可能存在multiple amino acid (or nucleotide) substiutions发生在任何给定的位置,Kimura距离矩阵为这种变化提供了一个模型。序列按照树的分支顺序被添加到多个序列比对中。在构建每棵树时,将其与第(1)阶段的树进行比较,该过程导致改进的渐进比对。
2.2.3、通过系统地对树进行划分,得到子集,迭代地细化指导树;删除树的边(分支)以创建一个双分区。
接下来,MUSCLE提取一对概要文件(多个序列对齐),并重新比对它们(执行概要文件比对)。该算法接受或拒绝新生成的比对基于sum-of-pairs score是否增加。系统地访问和删除树的所有边以创建双分区。这一迭代求精步骤非常迅速,而且早前已经证明可以提高多个序列对齐的精度。
三、Consistency-Based方法
在使用Feng-Doolittle方法进行渐进式比对时,会生成成对的比对分数并用于构建树。基于一致性的方法采用一种不同的方法,利用生成的多序列比对信息来指导成对比对。我们讨论了两个基于一致性的多序列比对程序:ProbCons 和T-COFFEE。MAFFT也包括一个基于一致性评分的迭代细化方法,而Ensembl程序Pecan(在“通过Ensembl分析基因组DNA比对”中讨论过)应用一致性方法对基因组DNA进行比对。一致性的概念是,对于序列x y z,如果残基xi与zk对齐,zk与yj对齐,那么xi应该与yj对齐。基于一致性的技术在关于多个序列的信息环境中,例如基于zk比对到xi及yi的知识调整xi到yi的得分。这种方法是独特的,因为它结合了来自多个序列的证据来指导成对比对的生成。基于一致性的方法通常会生成最终多序列比对,这些比对比基于标杆分析的渐进比对更精确。
3.1、T-COFFEE
T-COFFEE首先计算一个由pairwise alignments组成的库。默认情况下,这包括输入序列的所有可能的成对全局比对(使用Needleman-Wunsch算法),以及10个得分最高的局部比对。每对比对的残基都有一个权值。这些权重被重新计算,生成一个“扩展库”,作为位置特定的替换矩阵。然后,该程序通过渐进比对、创建距离矩阵、计算邻接引导树、使用动态程序明和从扩展库派生的替换矩阵来计算多序列比对。T-COFFEE包括一套相关的比对和评估工具。M-COFFEE(Meta-COFFEE)结合多达15种不同的多序列对齐方法的输出。这些包括T-COFFEE、ClustalW, MAFFT, MUSCLE和ProbCons。M-COFFEE采用一种基于一致性的方法来估计一致性比对比任何单独方法都更准确。
四、Structure-Based Methods
三级结构的演化比一级序列慢。通过将一个或多个成员中的蛋白三维结构信息包含其中,可以提高多序列比对的准确性。使您能够合并结构信息的程序包括PRALINE和T-COFFEE模块Expresso。当您在T-COFFEE网站上使用Expresso程序时,您提交了一系列序列(通常是FASTA格式),BLAST根据蛋白质数据库(PDB)自动搜索每个序列,并使用匹配(共享>60%氨基酸相似性)提供模板来指导对序列比对。结构信息也可以用来评估多个序列对齐后的准确性。这是在对已知结构的蛋白质家族进行基准测试研究(见下一节)中完成的。在另一种方法中,您可以在T-COFFEE包的iRMSD-APDB(“与蛋白质数据库分析比对”)服务器上分析结构信息并评估蛋白质多重序列比对的质量。
五、基准研究:方法、发现、挑战
生物信息学领域使用算法分析大范围应用中的数据,如配对比对、数据库搜索、测量RNA转录水平或预测蛋白质功能。通常有许多软件包可用来进行数据分析。我们怎么知道该相信哪一个呢?基准测试提供了一种重要的方法。我们可以得到一个“黄金标准”的正确答案,由可信的真正积极的关系组成,然后比较软件程序,客观地确定哪一个是最准确的。基准数据集可能包含多个序列比对的单独类别,例如具有不同长度、不同divergence、插入或删除的蛋白质不同长度和不同motifs的(indels)(如内部重复)。麦克卢尔et al(1994)对多序列比对进行了最早的基准研究之一,指出了全局比对算法而不是局部比对算法的优点。最近Aniba等人(2010)回顾了基准测试的概念,特别是在多序列比对方面。他们注意到基准数据库的下列品质:
相关性(Relevance):基准测试应该包括软件用户实际遇到的任务。
可解性( Solvability):任务不应该太容易(如蛋白质共享>50%相似性)或太难如共享有限序列和有限结构信息)
以上是关于序列比对的主要内容,如果未能解决你的问题,请参考以下文章