Predicting effects of noncoding variants with deep learning–based sequence model | 基于深度学习的序列模型预测非编码区
Posted leezx
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了Predicting effects of noncoding variants with deep learning–based sequence model | 基于深度学习的序列模型预测非编码区相关的知识,希望对你有一定的参考价值。
Predicting effects of noncoding variants with deep learning–based sequence model
这篇文章的第一个亮点就是直接从序列开始分析,第二就是使用深度学习获得了很好的预测效果。
This is, to our knowledge, the first approach for prioritization of functional variants using de novo regulatory sequence information.
该写的文章里都写得非常清楚了,再看不懂就是自己的问题了。
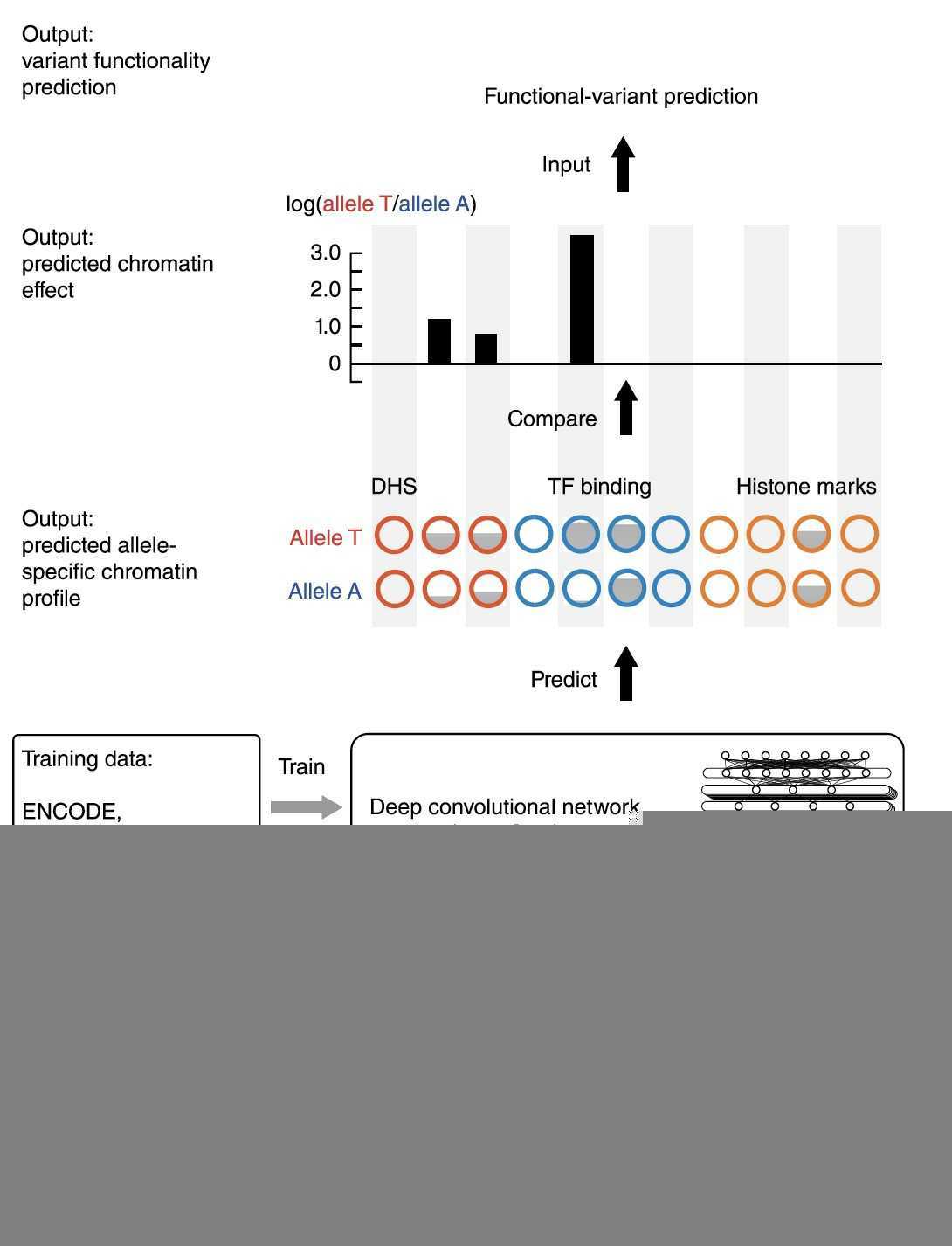
理解数据
List of all publicly available chromatin feature profile files used for training DeepSEA
ENCODE有哪些public的数据?本文中用了哪些数据来训练?
- DHS - wgEncodeAwgDnaseUniform
- TF binding - wgEncodeAwgTfbsUniform
- Histone marks - roadmap
To prepare the input for the deep convolutional network model, we split the genome into 200-bp bins. For each bin we computed the label for all 919 chromatin features; a chromatin feature was labeled 1 if more than half of the 200-bp bin is in the peak region and 0 otherwise.
这919 chromatin features都是些什么,怎么来的?(125 DNase features, 690 TF features, 104 histone features)
The 1,000-bp DNA sequence is represented by a 1,000 × 4 binary matrix, with columns corresponding to A, G, C and T. The 400-bp flanking regions at the two sides provide extra contextual information to the model.
训练集的上下游是加了400bp,但是训练的时候如何利用这额外的序列呢?
这里的input就很明确了:1000x4xN,然后每一个1000x4都对应了919个结果,也就是input的维度为1000,predict出来结果的维度为919.
这样,给定任何一个长度为1000的序列,就能得到一个919维度的关于chromatin feature的结果。
关于第一个model的实现就基本完成了。
那么第二个model呢?
For positive standards we used single-nucleotide substitution variants annotated as regulatory mutations in the HGMD professional version 2014.4 (ref. 17),
eQTL data from the GRASP 2.0.0.0 database with a P-value cutoff of 1 × 10−10 (ref. 1)
GWAS SNPs downloaded from the NHGRI GWAS Catalog on 17 July 2014 (ref. 18).
额外:Coding variants were filtered on the basis of the UCSC build hg19 knownGene track22.
SNP和eQTL的数据是如何被结合到当前的model里的?
To compute predicted chromatin effects of variants using the DeepSEA model, for each SNP, we obtained the 1,000-bp sequence centered on that variant based on the reference genome (specifically, the sequence is chosen so that the variant was located at the 500th nucleotide). Then we constructed a pair of sequences carrying either the reference or alternative allele at the variant position.
目的就是要预测每一个variant对919个chromatin feature的影响,此时我们就知道了任意一个序列的其中任意一个位点的SNP导致的919个chromatin feature差异结果。
然后这个如何与eQTL结合呢?
For each of the three variant types, HGMD single-nucleotide substitution regulatory mutations, eQTLs and GWAS SNPs, we trained a regularized logistic regression model, using the XGBoost implementation
这个分类模型是用来预测什么的呢?
The HGMD regulatory mutation model was trained with L1 regularization parameter 20 and L2 regularization parameter 2,000 for ten iterations. eQTL and GWAS SNP models were trained with L1 regularization parameter 0 and L2 regularization parameter 10 for 100 iterations.
可以看到这两个数据是分开train的,这些数据里面分别都有些什么信息?这个model到底想用什么来训练什么?SNP来训练出eQTL吗?
HGMD:Substitutions causing regulatory abnormalities are logged in with thirty nucleotides flanking the site of the mutation on both sides. The location of the mutation relative to the transcriptional initiation site, initiaton codon, polyadenylation site or termination codon is given.
GRASP: Genome-Wide Repository of Associations Between SNPs and Phenotypes 这里的eQTL应该是每一个SNP高度相关的基因表达的Rank。
第二个model介绍比较简单,因为很多人都在做这方面的工作,文章里也列出了很多个比较的工具。
但因为我没做过,所以暂时很难理解这部分的工作。
理解问题
无问题,不学习:
只关注non-coding区,那coding区的就不管了吗?coding区是直接对蛋白产生影响,同时还会对某些lncRNA的产生影响。需要扎实的基因组学知识。
预测variants的effect,这里的effect怎么直观理解?基因表达是effect吗?
这是一个什么样的深度学习模型?input的feature是什么?output的result是什么?
用了什么数据来训练这个模型?ENCODE和Roadmap
directly learns a regulatory sequence code from large-scale chromatin-profiling data, enabling prediction of chromatin effects of sequence alterations with single-nucleotide sensitivity.
该模型如何考虑了eQTL和疾病相关的variants?
关于non-coding variant你有什么理解?数量大,功能不明确,通过eQTL我们确实发现了一些与基因表达密切相关的位点。但显然,某个基因会有很多个eQTL位点,这些位点是如何具体影响到某个基因的表达的,我们不得而知。现在已经发现了eQTL位点在不同组织里具有不同的效应,说明了基因的选择性表达。由于中心法则的限制,变异能影响的有转录、转录调控和翻译。目前关于基因组序列的运作功能我们知之甚少,但它又是一切的源泉,所以最基本的变异带来的影响其实很复杂的,30亿个碱基,可能的变化组合太多了。从细节开始分析这些variant的功能的策略显然是行不通的。
我们的理解和认知极限,为什么我们对复杂系统的分析是如此的艰难?一个真实存在并高效运转的系统,我们却猜不透摸不着。
一个变异很好理解,找出细胞内所有与该位点直接相关的生物过程,再侦测后续如何发生连锁反应,最终会导致写什么结果!
基因组的变异是时刻在发生的,体细胞、生殖细胞,我们真正面对的是成千上万个突变,这些突变综合在一起是如何相互影响,导致最终的表型的变化的?这复杂度可就呈指数上升了,中间的过程可以很复杂很琐碎,我们人真正关注的还是最终的表型,生死和欲望。
no method has been demonstrated to predict with single-nucleotide sensitivity the effects of noncoding variants on transcription factor (TF) binding, DNA accessibility and histone marks of sequences.
A quantitative model accurately estimating binding of chromatin proteins and histone marks from DNA sequence with singlenucleotide sensitivity is key to this challenge.
Therefore, accurate sequence-based prediction of chromatin features requires a flexible quantitative model capable of modeling such complex dependencies—and those predictions may then be used to estimate functional effects of noncoding variants.基于序列的,可以考虑到更多复杂的情况,这比单纯的依赖现有的motif要更加准确。
We first directly learn regulatory sequence code from genomic sequence by learning to simultaneously predict large-scale chromatin-profiling data, including TF binding, DNase I sensitivity and histone-mark profiles.
sequence context
multilayer
share learned predictive sequence features
通俗来说:
这篇文章里有两个model,第一个是根据序列的特征来预测enhancer和promoter,然后再基于enhancer和promoter的epigenome的内容来预测eQTL。
model1:TF binding, DNase I sensitivity and histone-mark profiles
文章晦涩难懂可以直接先跑下代码试下:
代码在这:http://deepsea.princeton.edu/job/analysis/create/
三种可选的input文件:
FASTA
>[known_CEBP_binding_increase]GtoT__chr1_109817091_109818090 GTGCCTCTGGGAGGAGAGGGACTCCTGGGGGGCCTGCCCCTCATACGCCATCACCAAAAGGAAAGGACAAAGCCACACGC AGCCAGGGCTTCACACCCTTCAGGCTGCACCCGGGCAGGCCTCAGAACGGTGAGGGGCCAGGGCAAAGGGTGTGCCTCGT CCTGCCCGCACTGCCTCTCCCAGGAACTGGAAAAGCCCTGTCCGGTGAGGGGGCAGAAGGACTCAGCGCCCCTGGACCCC CAAATGCTGCATGAACACATTTTCAGGGGAGCCTGTGCCCCCAGGCGGGGGTCGGGCAGCCCCAGCCCCTCTCCTTTTCC TGGACTCTGGCCGTGCGCGGCAGCCCAGGTGTTTGCTCAGTTGCTGACCCAAAAGTGCTTCATTTTTCGTGCCCGCCCCG CGCCCCGGGCAGGCCAGTCATGTGTTAAGTTGCGCTTCTTTGCTGTGATGTGGGTGGGGGAGGAAGAGTAAACACAGTGC TGGCTCGGCTGCCCTGAGGTTGCTCAATCAAGCACAGGTTTCAAGTCTGGGTTCTGGTGTCCACTCACCCACCCCACCCC CCAAAATCAGACAAATGCTACTTTGTCTAACCTGCTGTGGCCTCTGAGACATGTTCTATTTTTAACCCCTTCTTGGAATT GGCTCTCTTCTTCAAAGGACCAGGTCCTGTTCCTCTTTCTCCCCGACTCCACCCCAGCTCCCTGTGAAGAGAGAGTTAAT ATATTTGTTTTATTTATTTGCTTTTTGTGTTGGGATGGGTTCGTGTCCAGTCCCGGGGGTCTGATATGGCCATCACAGGC TGGGTGTTCCCAGCAGCCCTGGCTTGGGGGCTTGACGCCCTTCCCCTTGCCCCAGGCCATCATCTCCCCACCTCTCCTCC CCTCTCCTCAGTTTTGCCGACTGCTTTTCATCTGAGTCACCATTTACTCCAAGCATGTATTCCAGACTTGTCACTGACTT TCCTTCTGGAGCAGGTGGCTAGAAAAAGAGGCTGTGGGCA >[known_FOXA2_binding_decrease]ReferenceAllele__chr10_23507864_23508863 CTTCTTTTTATCTCTTAACTAACTTACAATTTCTTACGTGATTTTAAAACTTGTTTTTCTATTTAAAACAACAGGGGCAA CTGAACTTCACTTTCAAACAATATTTATTTCTATAAATCAGTGCAAAACATACTTATTGAAAATATATCTTGGGTCCAAG GCTTCAAAGGGTAAAAAGAAAGATTTTAAATTATATCTAATATGTTACAATTGTTCTGTCCTTTAAAAACCTTTTCAGAT CACCCCCTGGATGATTCTTCCCTAGAAGTCTCAGAGAATTAACAACACAATGTAATCTAGGTTTAAATTTGGGTTTCTCC TGTGTTTCAGATACTGATGTTTGAGCTTTCTCTTCCTGACAAGCCACTTAAAGAGTCACTGTTACTTTGAGGTTTTATCT GTAAGATTCGTGTCTTTTGGGCTCATTAAGAACATTTCCAAAGATTACAATGTCAATAGCACCTAATTACTGGACTGTGA GAAAGGTCTTCTTGAGTACATAAAATCTGTGGCAGTGCACAGTACACAATGGGCAGCTCAGATCCCAAATTTTATCACAA GTAAGTAGCAAACAAATTAATAATGTTACCTGTGCTCTCTTGGATAATTACTACTGCATAAAAACTGCTTTGAAATGTTG CAGATAGTATTGTACCTCATTTTTTTAATCCCCTTAGAGTAACAAGGATTTATTTGTCTCAAACTTTCTATGTTGCATGC ACCACTTGACTTTCTTGTTCTGTTTAGAATTTTTAGAACTTGCAACATAACAAAAAATCATTTTTAACCAGCCTAGGAAG GACATATCACCTGATGTAACATTATTTTAAATTATATTTTGTATTTTACTTTACTCTTTTCAAAACATATACTGTATGTT TTGATACTATTGCTAGATTTTATTTTTTACTTATGCCTGGTAGAAAATCAGCTATTAAAGAAGCAGAGGAGGCTGGACAC AGTGGTTCATGTCTGTAATCGCTAGCACTTTGAAAGAGTA >[known_GATA1_binding_increase]TtoC__chr16_209210_210209 GGGCTTAGACAGAGGAGGGGAGGATTCAGATTTTAAATGGGTTGGCCACTGTAGGTCTATTAACGTGGTGACATTTGAGG GAGTGGCAATACTAGGGAAGGGGCTTCAGGGGAGTGGCCAGGAGCTAGGGATAGAGGGAGGGAGGACAGGAGGCCTTGTC TGTCTTTTCCTCCATATGTAAGTTTCAGGAGTGAGTGGGGGGTGTCGAGGGTGCTGTGCTCTCCGGCCTGAGCCTCAGGA AGGAAGGGCAGTAGTCAGGGATGCCAGGGAAGGACAGTGGAGTAGGCTTTGTGGGGAACTTCACGGTTCCATTGTTGAGA TGATTTGCTGGAGACACACAGATGAGGACATCAAATACATCCCTGGATCAGGCCCTGGGGCCTGAGTCCGGAAGAGAGGT CTGTATGGACACACCCATCAATGGGAGCACCAGGACACAGATGGAGGCTAATGTCATGTTGTAGACAGGATGGGTGCTGA GCTGCCACACCCACATTATCAGAAAATAACAGCACAGGCTTGGGGTGGAGGCGGGACACAAGACTAGCCAGAAGGAGAAA GAAAGGTGAAAAGCTGTTGGTGCAAGGAAGCTCTTGGTATTTCCAATGGCTTGGGCACAGGCTGTGAGGGTGCCTGGGAC GGCTTGTGGGGCACAGGCTGCAAGAGGTGCCCAGGACGGCTTGTGGGGCACAGGTTGTGAGAGGTGCCCTGGACGGCTTG TGGGGCACAGGCTGTGAGAGGTGCCCAGGACGGCTTGTGGGGCACAGGCTGTGAGGGTGCCCGGGACGGCTTGTGGGGCA CAGGTTGTGAGAGGTGCCCGGGACGGCTTGTGGGGCACAGGTTTCAGAGGTGCCCGGGACGGCTTGTGGGGCACAGGTTG TGAGAGGTGCCCGGGACGGCTTGTGGGACACAGGTTGTGAGAGGTGCCTGGGACGGCTTGTGGGGCACAGGCTGTGAGGG TGCCTGGGACGGCTTGTGGGGCACAGGTTGTGAGAGGTGC >[known_FOXA1_binding_increase]CtoT_chr16_52598689_52599688 GGCTCAAGCAGTCCTCCCATCTAGGCTTCCCAAAATGCTGGGATTACAGACATGAGCCACTGCACCCAGCCACAAAGATA ACCTAAAGATGTGTTTACTTTGACCCAGGCAGTAGTTTAAAAAAGTTTTAATTTGTTGTTCACATTTAAAAACTGGACAA TTTCTACATAAAAATCTGAATTACTCATGTCTCTTAAAAAAATAACATCTAGCAATGGTAGGCCCACATTCCTTCCTGAA AATAATTAGCTGGGAAAGAGTAGGGACTGACCCCTTTAGACACGGTATAAATAGCATGGGAGTTGATCAGTAAATATTTG CTGAATGAAAGAATACATGAATGAAAAGTCAGAGCCCTATAGGTCAGCATGGACGGCGGTAAAGGAACCTGGCTGAGCCT GAAAGAGAATGTGATCTAAGATTAAATCCAGGATATGCTGGTAAATGTTTAACAGCCAACTCTTTGGGGAGGAAAAAAGT CCCAATTTGTAGTGTTTGCTGATTATTGTGATGTAAATACTCCCATCATGACCAATTTCAAGCTACCAACATGCTGACAC TGAACTTGGAGTTGGAAGGAGATGAACAGGCATAATCAGGTCTCGTGAGATGGCCCAAGCCGGCCCCAGCACTCCACTGT TATATATGAGGCTAGAATTACTACATAACTGGAATAGCAACTTTCTGGACCATATGCCTGGAACACAGCAGGTGCTGAAT AAATGTTTGTTGATCCAGGAACTGACTGTGTTGAAGCCCACAGATGGGAAATCAGTAGAAGGCAGGTAAGAGTAAAAAGA AGGGCAGAGAATTGGGGGTACAGACCCCTGAACCATAAGTCAGAGGAATGTTGTACATGTTTTCAGATCCCTCACTGGTC AAATGAAGGCAAAGGGTTAGATCTCTCCAAATCTTTAGAGGGACATGATGTAACTCCATTAAGTAACTCAGTGATTTTCA ACATTAAAAAGTGTAATTATCTTTTCAAACTAAATATTAC
VCF
chr1 109817590 [known_CEBP_binding_increase] G T chr10 23508363 [known_FOXA2_binding_decrease] A G chr16 52599188 [known_FOXA1_binding_increase] C T chr16 209709 [known_GATA1_binding_increase] T C
BED
chr1 109817090 109818090 . 0 * chr10 23507863 23508863 . 0 * chr16 52598688 52599688 . 0 * chr16 209209 210209 . 0 *
网页结果:
因为motif的binding是一个概率学的东西,所以理论上单个碱基的改变会导致TF在该motif上binding的增加和减少,网页里面就是想预测出来某个特定的variant会导致哪些TF的binding出现显著差异。
但是还是不懂图里面的chromatin feature和log2FC是什么意思!
如何理解这个Functional significance score,以及整个table?
关于模型的训练:
数据
ENCODE and Roadmap Epigenomics data were used for labeling and the HG19 human genome was used for input sequences. The data is splitted to training, validation and test sets. The genomic regions are splitted to 200bp bins and labeled according to chromatin profiles. We kept the bins that have at least one TF binding event (note that TF binding event is measured by any overlap with a TF peak, not the >50% overlap criterion used for labeling).
DEPENDENCIES
1. Install CUDA driver. A high-end NVIDIA CUDA compatible graphics card with enough memory is required to train the model. I use Tesla K20m with 5Gb memory for training the model.
2. Installing torch and basic package dependencies following instructions from
http://torch.ch/docs/getting-started.html
You may need to install cmake first if you do not have it installed. It is highly recommended to link against OpenBLAS or other optimized BLAS library when building torch, since it makes a huge difference in performance while running on CPU.
3. Install torch packages required for training only: cutorch, cunn, mattorch. You may install through `luarock install [PACKAGE_NAME]` command. Note mattorch requires matlab. If you do not have matlab, you may try out https://github.com/soumith/matio-ffi.torch and change 1_data.lua to use matio instead (IMPORTANT: if you use matio, place remove the ":tr\\
anspose(3,1)" and "transpose(2,1)" operation in 1_data.lua. The dimesions have been correctly handled by matio.).
这种必须要用显卡了,N卡,用的CUDA平台。二手的Tesla K20m with 5Gb memory大概两千多。
Usage Example
th main.lua -save results
The output folder will be under ./results . The folder will inlcude the model file as well as log files for monitoring training progress.
You can specify various parameters for main.lua e.g. set learning rate by -LearningRate. Take a look at main.lua for the options.
Short explanation of the code: 1_data.lua reads the training and validation data; 2_model.lua specify the model; 3_loss.lua specify the loss function; 4_train.lua do the training.
可以看到训练model的任务是用lua语言来写的。
关于Sequence Profiler功能
"in silico saturated mutagenesis" analysis for discovering informative sequence features within any sequence.
预测所提供序列上每一个位点发生各种突变时的log2FC的变化情况。
参考:
zhoujian最新的文章:Nature Genetics | 深度学习揭示大量自闭症相关非编码区突变
以上是关于Predicting effects of noncoding variants with deep learning–based sequence model | 基于深度学习的序列模型预测非编码区的主要内容,如果未能解决你的问题,请参考以下文章