QIIME2使用方法
Posted afeiyuanda
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了QIIME2使用方法相关的知识,希望对你有一定的参考价值。
激活qiime2的执行环境:source activate qiime2-2019.4
如何查看conda已有的环境:conda info -e
以下分析流程参考:https://docs.qiime2.org/2019.4/tutorials/qiime2-for-experienced-microbiome-researchers/
1、数据准备
现在我们常用的就是这种格式的数据,每个样品一对数据文件
wget -O "casava-18-paired-end-demultiplexed.zip" "https://data.qiime2.org/2019.4/tutorials/importing/casava-18-paired-end-demultiplexed.zip"
下载解压后,文件夹中文件如下:
2、将数据转换为qza格式(qiime新定义的自己的格式类型,有点编程中对象的含义)
qiime tools import --type ‘SampleData[PairedEndSequencesWithQuality]‘ --input-path casava-18-paired-end-demultiplexed --input-format CasavaOneEightSingleLanePerSampleDirFmt --output-path demux-paired-end.qza
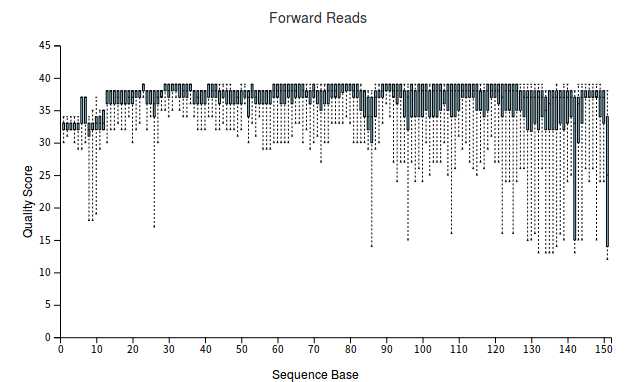
3、查看数据质量
qiime demux summarize --i-data demux-paired-end.qza --o-visualization demux-summary-1.qzv
用以下命令查看结果:
qiime tools view demux-summary-1.qzv
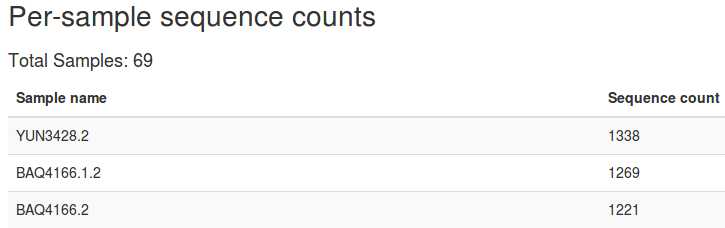
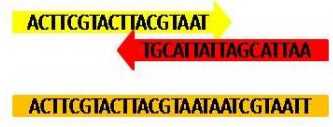
4、双端序列合并成单端
qiime vsearch join-pairs --i-demultiplexed-seqs demux-paired-end.qza --o-joined-sequences demux-joinded.qza
5、查看对merge后的数据质量情况
qiime demux summarize --i-data demux-joinded.qza --o-visualization demux-summary-merged.qzv
qiime tools view demux-summary-merged.qzv
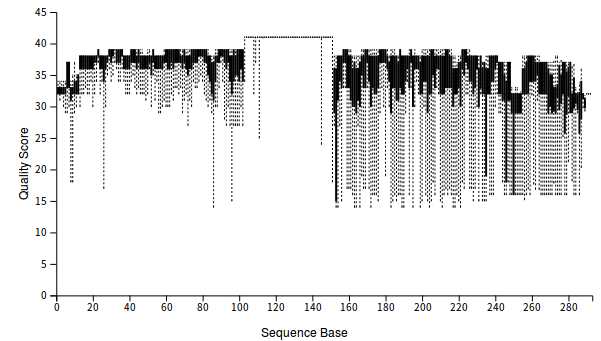
4、对数据进行剪切
双端:
qiime dada2 denoise-paired --i-demultiplexed-seqs demux-paired-end.qza --p-trim-left-f 13 --p-trim-left-r 13 --p-trunc-len-f 150 --p-trunc-len-r 150 --o-table table.qza --o-representative-sequences rep-seqs.qza --o-denoising-stats denoising-stats.qza
单端:
qiime dada2 denoise-single --i-demultiplexed-seqs demux-joinded.qza \\ #输入应该也是序列,不能是joined对象
--p-trim-left 13 \\
--p-trunc-len 150 \\
--o-table table.qza --o-representative-sequences rep-seqs-merged.qza --o-denoising-stats denoising-stats-merged.qza
https://forum.qiime2.org/t/demultiplexing-and-trimming-adapters-from-reads-with-q2-cutadapt/2313
以上是关于QIIME2使用方法的主要内容,如果未能解决你的问题,请参考以下文章