python学习——通过命令行参数根据fasta文件中染色体id提取染色体序列
Posted caicai2019
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了python学习——通过命令行参数根据fasta文件中染色体id提取染色体序列相关的知识,希望对你有一定的参考价值。
提取fasta文件genome_test.fa中第14号染色体的序列,其内容如下:
>chr1 ATATATATAT >chr2 ATATATATATCGCGCGCGCG >chr3 ATATATATATCGCGCGCGCGATATATATAT >chr4 ATATATATATCGCGCGCGCGATATATATATCGCGCGCGCG >chr5 ATATATATATCGCGCGCGCGATATATATATCGCGCGCGCGATATATATAT >chr6 ATCGATGCAGCATG >chr7 TATCGCGCGCGCGATATAT >chr8 ATATCGCGCGCGCGATATATATATCGCG >chr9 ATCGCGCGCGCGATATATATATCGCG >chr10 GCGCGCGATATAT >chr11 CGCGATATATATATC >chr12 ATATATCGCGCGCGCGATATAT >chr13 ATATATCGCGCGCGCGATATATGCGATATATATATC >chr15 ATATATGCGAT >chr14 GCGCGCGCGATATATGCGAT >chr16 GCGATATATGCGATATATATATC >chr17 GCGCGCGCGATATATATATCGCGCGCGCGATATATATAT >chr18 GCGCGCGCGATATATATATCGCGCGCGCGATATATATATATATGCGATATATATATC >chr19 ATATGCGATATATATATCGCGCGCGCGATATATATATCGCGCGCGCGATATATATATATATGCGA >chr20 TATGCGATATATATATCGCGCGCGCGATATATATATCGCGCGCGCGATATATATATATATGCGA >chr21 TATATCGCGCGCGCGATATATATATCGCGCGCGCGATATATATATATATGCGA >chr22 ATATATATCGCGCGCGCGATATATATATATATGCGA >chrX CGCGCGCGATATATATATATATGCGA >chrY CGCGCGCGATATATATATATATGCGACGCGCGCGATATATATATATATGCGACGCGCGCGATATATATATATATGCGA
用python以及命令行参数实现
新建.py文件“”GetSeqFromChrID.py”,
python脚本如下:
1 import argparse 2 3 def read_fasta(input): 4 5 with open(input, ‘r‘) as f: 6 fasta = {} 7 for line in f: 8 line = line.strip() 9 if line[0] == ‘>‘: 10 header = line[1:] 11 else: 12 sequence = line 13 fasta[header] = fasta.get(header, ‘‘) + sequence 14 15 return fasta 16 17 18 if __name__ == ‘__main__‘: 19 # read arguments 20 parser = argparse.ArgumentParser(description="this program is used to extract a single " 21 "sequence from genome") 22 parser.add_argument(‘--input‘, ‘-i‘, 23 type=str, 24 help=‘input file in fasta format‘) 25 parser.add_argument(‘--output‘, ‘-o‘, 26 type=str, 27 help=‘output file‘) 28 parser.add_argument(‘seq_id‘, 29 type=str, 30 help=‘sequence id‘) 31 args = parser.parse_args() 32 33 fasta = read_fasta(args.input) 34 with open(args.output, ‘w‘) as f: 35 f.write(‘>{:s}\\n{:s}\\n‘.format(args.seq_id,fasta.get(args.seq_id, ‘can not found this sequence‘)))
命令行参数输入如下:红色字体是输入部分
1 (base) e:\\15_python\\DEBUG>python GetSeqFromChrID.py -i genome_test.fa -o chr14.fa chr14
结果如下:
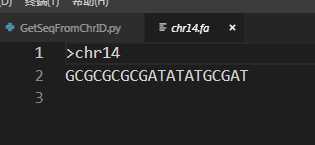
以上是关于python学习——通过命令行参数根据fasta文件中染色体id提取染色体序列的主要内容,如果未能解决你的问题,请参考以下文章