同源建模方法整理
Posted xiaojikuaipao
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了同源建模方法整理相关的知识,希望对你有一定的参考价值。
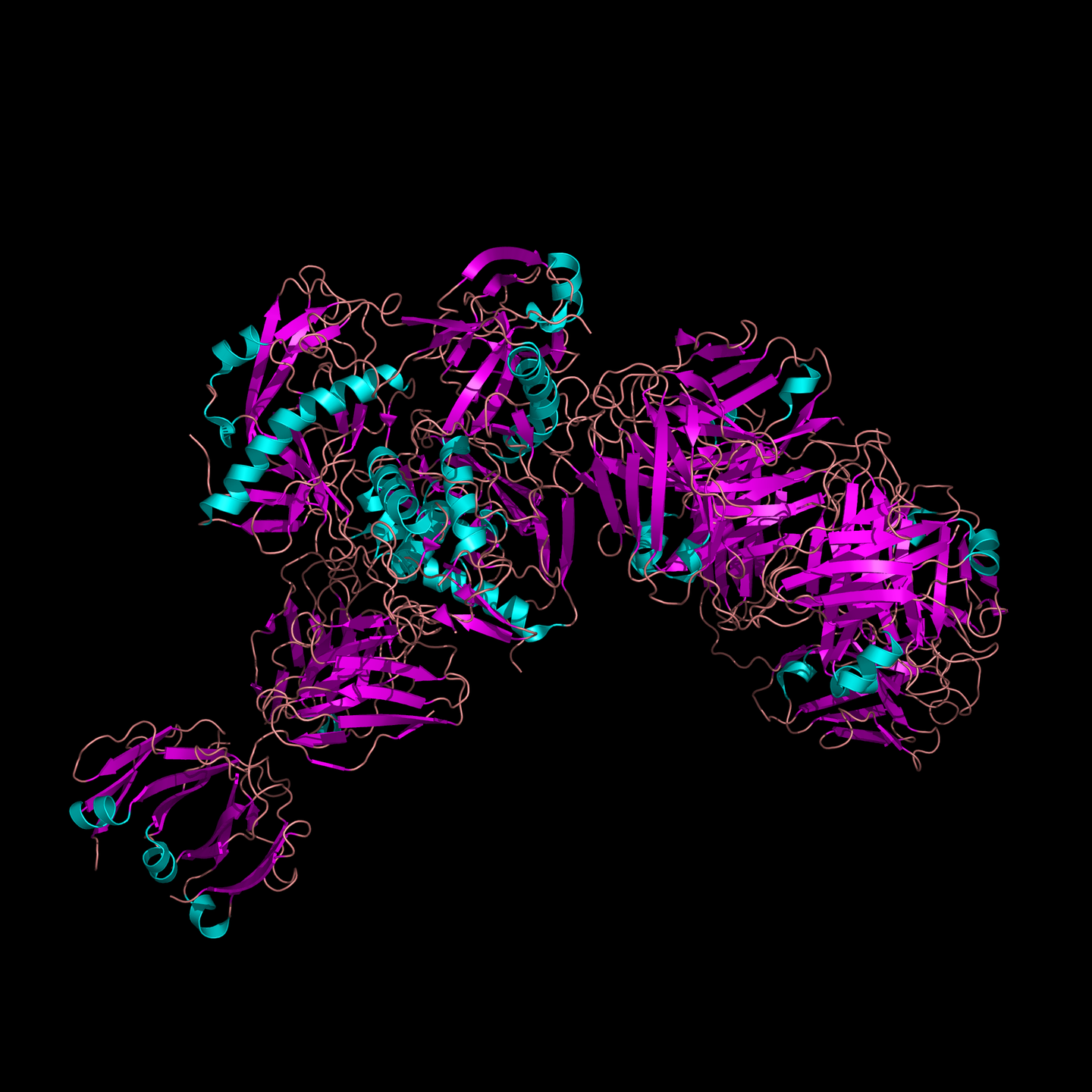
同源建模方法整理
之前一直是使用Swiss model(http://swissmodel.expasy.org/interactive)做同源建模,这个服务器很简单,只要输入自己目的蛋白的序列即可,或者你自己提供模板,再输入蛋白序列,界面友好,但是它有几点不足之处,比如当你用蛋白A来模建蛋白B的时候,A序列比B序列短,那么构建出来的B就会缺少氨基酸,也就是说它不能自动补充那几个少的氨基酸,同源建模的理论前提是模板和目的蛋白的序列相似度要高于30%,对于没有高度相似模板的蛋白也是无法用这个服务器的。最近我需要构建一个蛋白,而这个蛋白没有序列相似的模板,那么这种情况就需要从头建模的方法,我使用的是I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/)。它的好处是你可以自己设置二硫键连接方式,二级结构,模板,从头建模,意味着从头到尾,每个氨基酸都不缺少,但是它构建的时间较长,往往需要半天到一天出结构,且不能一个账号同时提交多个任务,只能等一个任务结束后再提交下一个。
该文是为那些未做过同源建模的同学提供,对于已非常熟悉该方法的,就不需要花时间看这篇文章了。当你拿到一个多肽或蛋白序列A,要构建它的结构,可依照下面的步骤:
1. 搜索是否有同源蛋白的结构解析
用Blastp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome),搜索protein data bank数据库(一个收集了所有结构解析了的多肽及蛋白的数据库)里是否有A的同源序列
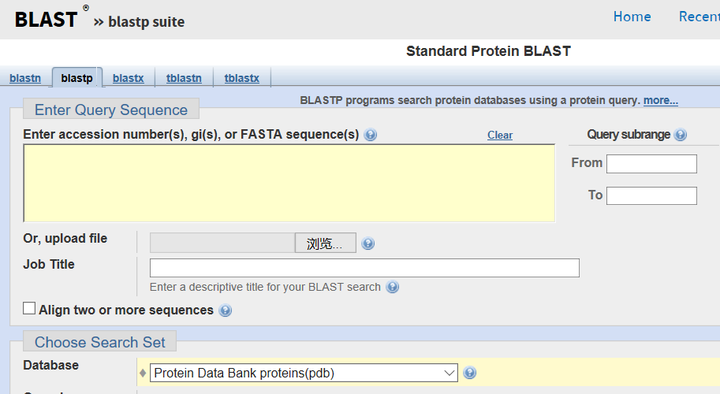
2 建模
2.1 有同源序列且序列相似度高于30%,且同源序列的长度比A长
那么就选择Swiss model (http://swissmodel.expasy.org/interactive)进行同源模建
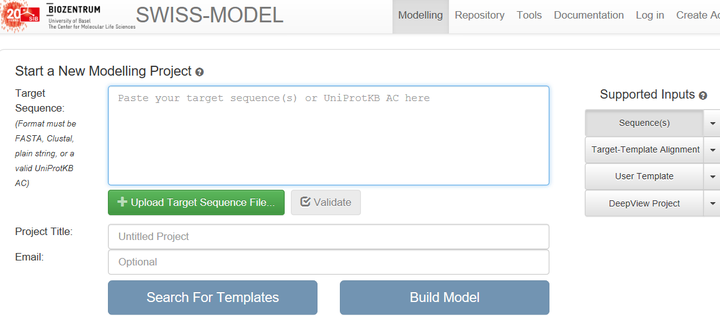
2.2 无同源序列,或者同源序列比A短很多
只能选择从头模建的方法,这里推荐I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/),因为我只用过这个服务器,界面也很友好,需要简单注册一个账号
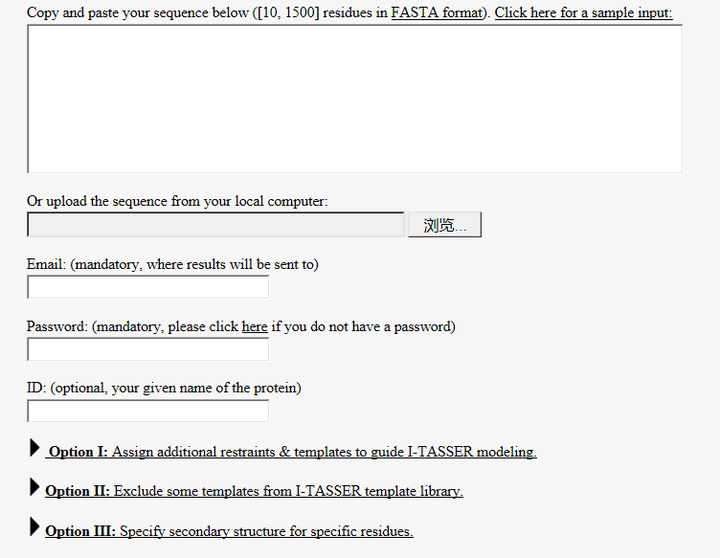
(1) 在OptionI中可设置二硫键的连接方式(点击Explanation里面有详细的设置氨基酸连接的书写方式):

(2) 在OptionIII中可设置二级结构,哪些氨基酸是α-helix,哪些是β-sheet等(点击Explanation里面有详细的书写方式)
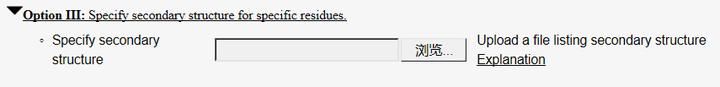
当然还有其它选项,如可自行添加template等,按照个人需要去设置
3 结构优化
3.1 调节二硫键
有的二硫键距离靠近但未连接上,可用spdbv软件将二硫键连接上,方法如下:
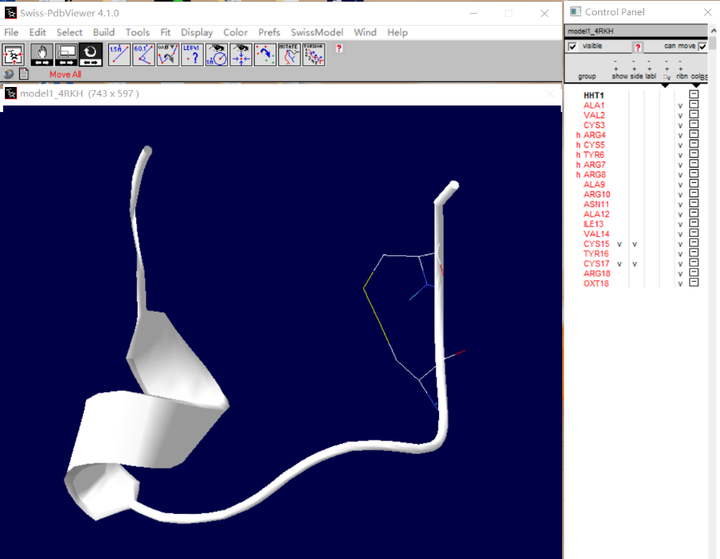
(1) 打开spdbv软件,将待分析结构拖入软件中
(2) 如图要连接这两个半胱氨酸,将两者侧链展示出来,如图:
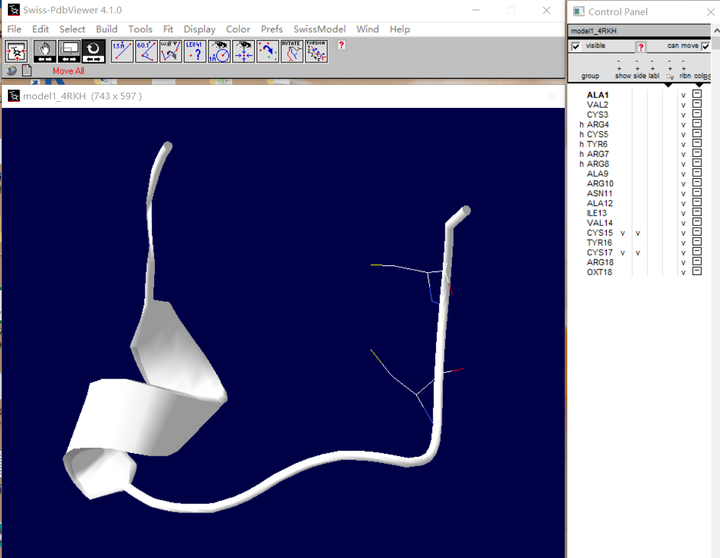
(3) 旋转二硫键
A. 先点击一下如下红色圈圈中的图标,再点击要旋转的半胱氨酸的侧链原子(黄色圈圈中)
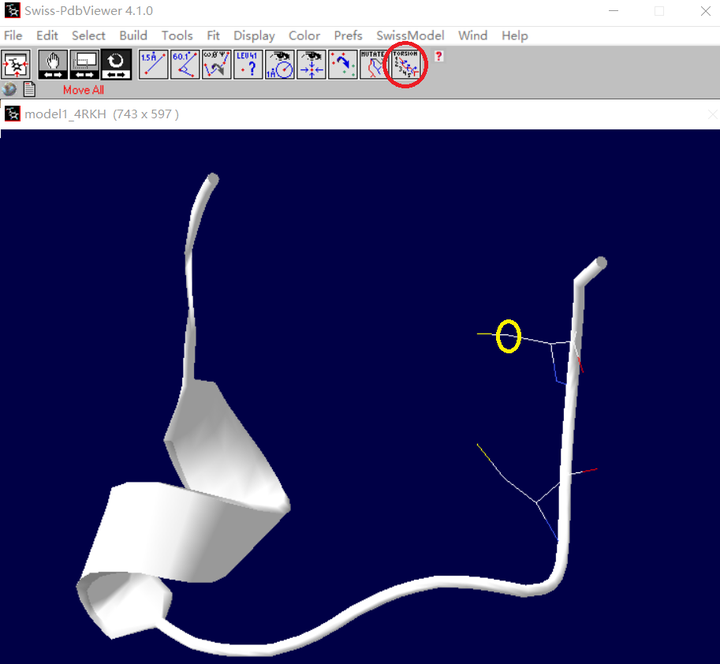
B. 然后会出现如下这样的黑色箭头(红色框框出),按这些箭头即可调节侧链的位置,使两者侧链靠近
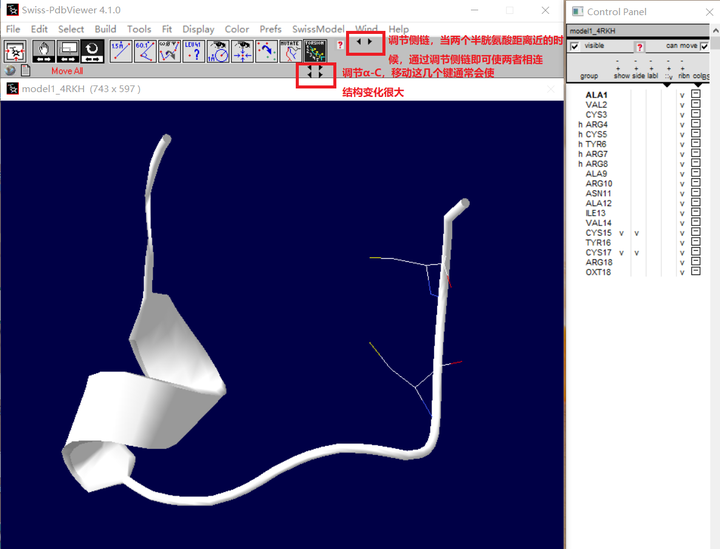
C. 能量最小化
选中所有的氨基酸(左键在control panel框里全选所有的氨基酸,如氨基酸被选中,在control panel框中会变为红色)
选中后,选择最上面一排工具栏中的Tools—energy minimazition即完成能量最小化
保存结构
File—Save—Current layer
3.2 能量最小化
方法较多,可使用gromacs这类动力学软件进行能量最小化,也可简单使用spdbv软件进行能量最小化。
其它结构的优化按照个人需求。
4 展示结构
我常用的展示蛋白结构的软件是pymol和molmol,pymol界面友好,更容易操作,molmol需要差一些命令,但其做出的图也很好看,各有优缺点,我是两者互补着用。它们常用的功能如下:
4.1 pymol
原子叠加(用于结构拼接时候);
设定二级结构原件(可自行定义某段氨基酸的二级结构);
连接化学键,如生成缺失的酰胺键;
原子间距离的计算和展示;
4.2 Molmol
展示二硫键;
自动连接缺失的酰胺键;
以上是关于同源建模方法整理的主要内容,如果未能解决你的问题,请参考以下文章
计算机辅助药物设计(AI)-分子对接-同源建模-药物筛选-先导化合物-机器学习药物发现
1.27 Java学习系列(二十七)UML建模的理解和图形整理