如何使用ChromHMM鉴定染色质状态
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了如何使用ChromHMM鉴定染色质状态相关的知识,希望对你有一定的参考价值。
参考技术A ChromHMM是基于ChIP-seq组蛋白数据检测染色质状态的工具。基因组区域特定的组蛋白修饰或修饰的组合具有特定的功能。
特定标记的区域和作用:
获取上述标记区域的方式通常是call peak, 但是,理想的情况下需要获取多个组蛋白标记的整合结果。ChromHMM是一个基于HMM模型的用于检测染色质状态的java软件。
Chromatin states的定义基于组蛋白修饰的不同组合和对应的不同功能区域。
其目的是将基因组区分为具有生物学功能的区域或片段。
以下为染色质segment的案例:
一、如何使用ChromHMM
(1)使用环境: java
(2)reads比对后的文件
如果自己进行分析,还需要: 比对软件和bedtools
二、使用流程如下:
1、测序获取reads
2、 reads比对到参考基因组
3、 转换aligned reads为BED格式
4、 创建Binned和Binarized tracks
5、 训练模型
6、 infer the states
7、 解读
三、具体操作步骤
(1) Alignment
多种短序列比对软件可用,BWA、Bowtie2等
(2) 比对reads转换为BED格式
bedtools bamtobed -i sample.bam > sample.bed
(3) 创建Binned and Binarized Tracks
java –mx4000M –jar ChromHMM.jar BinarizeBed –b 200 CHROMSIZES/hg18 cellmarkfiletable.txt SAMPLEDATA_HG18
其中,cellmarkfiletable.txt记录多个样本的修饰BED数据,例如:
cell1 mark1 cell1_mark1.bed cell1_control.bed
cell1 mark2 cell1_mark2.bed cell1_control.bed
cell2 mark1 cell2_mark1.bed cell2_control.bed
cell2 mark2 cell2_mark2.bed cell2_control.bed
(4)训练模型并segment基因组
java -mx1600M -jar ChromHMM.jar LearnModel SAMPLEDATA_HG19 OUTPUTSAMPLE 10 hg19
ChromHMM的输出:
生成html报告 webpage_N.html (N是状态)
包含信息如下:
1. Model learned: transi-on and emission parameters
2. Enriched func-onal categories
3. BED files to visualize the segmenta-on
可视化segmentation:
Genome Browser: https ://genome.ucsc.edu/
IGV: https ://www.broadins-tute.org/igv/
其他参考 :
1. Segway: https ://pmgenomics.ca/ho ffmanlab/proj/segway/
2. Spectacle: https ://github.com/jiminsong/Spectacle
ATAC-seq
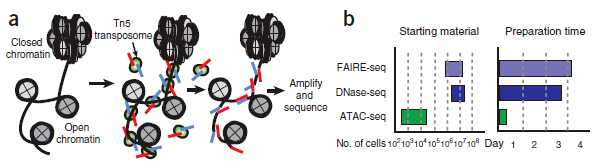
ATAC-seq(Assay for Transposase-Accessible Chromatin with high throughput sequencing) 是2013年由斯坦福大学William J. Greenleaf和Howard Y. Chang实验室开发的用于研究染色质可及性(通常也理解为染色质的开放性)的方法, 原理是通过转座酶Tn5容易结合在开放染色质的特性,然后对Tn5酶捕获到的DNA序列进行测序。
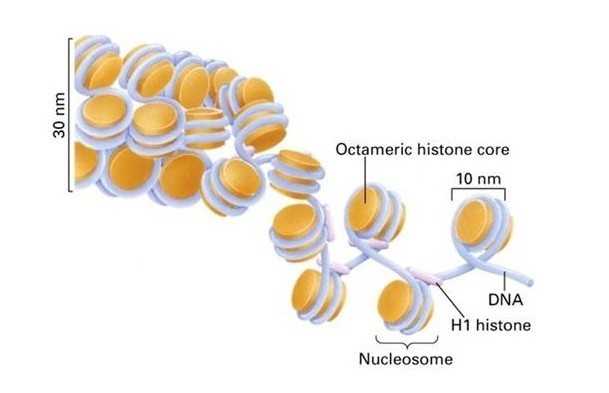
真核生物的核DNA并不是裸露的,而是与组蛋白结合形成染色体的基本结构单位核小体,核小体再经逐步的压缩折叠最终形成染色体高级结构(如人的DNA链完整展开约2m长,经过这样的折叠就变成了纳米级至微米级的染色质结构而可以储存在小小的细胞核)。而DNA的复制转录是需要将DNA的紧密结构打开,从而允许一些调控因子结合(转录因子或其他调控因子)。这部分打开的染色质,就叫开放染色质,打开的染色质允许其他调控因子结合的特性称为染色质的可及性(chromatin accessibility)。因此,认为染色质的可及性与转录调控密切相关。
开放染色质的研究方法有ATAC-seq以及传统的DNase-Seq及FAIRE-seq等,ATAC-Seq由于所需细胞量少,实验简单,可以在全基因组范围内检测染色质的开放状态,目前已经成为研究染色质开放性的首选技术方法。
Nat Methods, 2013. doi: 10.1038/nmeth.2688. Epub 2013 Oct.
ATAC-Seq与ChIP-Seq的异同
ATAC-Seq是全基因组范围内检测染色质的开放程度,可以得到全基因组范围内的蛋白质可能结合的位点信息,用此方法与其他方法结合筛查感兴趣的特定调控因子;
但是ChIP-Seq是明确知道感兴趣的转录因子是什么,根据感兴趣的转录因子设计抗体去做ChIP实验拉DNA,验证感兴趣的转录因子是否与DNA存在相互作用。
ATAC-Seq、ChIP-Seq、Dnase-Seq、MNase-Seq、FAIRE-Seq整体的分析思路一致,找到富集区域,对富集区域进行功能分析。
ChIP-Seq是揭示特定转录因子或蛋白复合物的结合区域,实际是研究DNA和蛋白质的相互作用,利用抗体将蛋白质和DNA一起富集,并对富集到的DNA进行测序。
DNase-Seq、ATAC-Seq、FAIRE-Seq都是用来研究开放染色质区域。DNase-Seq是用的DNase I内切酶识别开放染色质区域,而ATAC-seq是用的Tn5转座酶,随后进行富集和扩增;FAIRE-Seq是先进行超声裂解,然后用酚-氯仿富集。
MNase-Seq是用来鉴定核小体区域。
来源:https://www.jianshu.com/p/87bc2002e82c
source: https://www.jianshu.com/p/9c97734fe702
一文了解ATAC-seq
Assay for Transposase-Accessible Chromatin with highthroughput sequencing(ATAC-Seq)即利用转座酶探究可接近性染色质高通量测序技术。通俗来说就是利用转座酶来获取开放性染色质,再通过高通量测序及生物信息学分析来挖掘相关基因信息,以此探究生物学相关问题。
Q:为什么研究染色质开放区域?
A:染色质分为常染色质和异染色质,在结构上常染色质折叠压缩程度低,处于伸展状态,DNA复制,基因转录都发生在DNA的致密高级结构变为松散的状态;这部分打开的染色质,就叫开放染色质(open chromatin)。而打开的染色质,就有足够的区域允许一些调控蛋白(比如转录因子和辅因子)过来与之相结合。而染色质的这种特性,就叫做染色质的可接近性(chromatin accessibility)。通过研究细胞特定状态下开放的染色质区域可以在DNA水平上了解其转录调控。
Q:如何寻找开放的染色质区域?
A:传统使用的的实验方法主要是有MNase-seq和DNase-seq ,这两种实验方法的主要思路是:染色质变得开放,就意味着DNA和组蛋白的聚集程度降低,就会有一部分DNA暴露出来。而一旦失去了蛋白质的保护,这部分DNA就可以被DNA酶(MNase或DNase I)所切割。然后,我们再把切割完的DNA拿来测序,和已知的全基因组序列相比较,就能发现被切割的是哪些序列,没有被切掉的基因序列又在哪里,就知道开放的染色质区域在哪里了。不过,这两个方法有明显的缺陷,即耗时费力与重复性差。虽然FAIRE-seq 不依赖酶和抗体,但其检测背景较高,测序信噪比低,甲醛交联时间不好把握等缺陷,限制其使用范围。
一文了解ATAC-seq
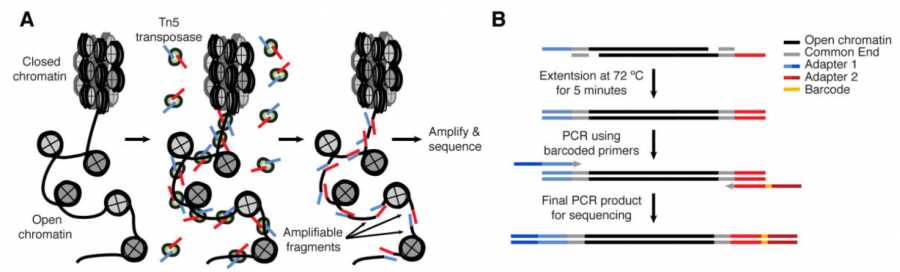
图1 ATAC-seq原理示意图
Q:有什么新技术方法来研究开放染色质?
A:新推出的ATAC-seq利用Tn5转座酶(DNA转座,是一种把DNA序列从染色体的一个区域搬运到另外一个区域的现象,这一过程就由转座酶参与完成。Tn5转座酶:“标签片段化工具”,Tn5转座体可将其衔接子负载整合到可接近的染色质区域,而空间位阻较不可接近的染色质使得转座不可能发生。)人为将将携带已知DNA序列标签的转座复合物,加入到细胞核中,再利用已知序列的标签进行PCR建库测序,就知道哪些区域是开放染色质了。ATAC-seq出来的结果,和传统方法出来的结果具有很强的一致性,同时也和ChIP-seq有较高的吻合程度。而相比较而言,ATAC-seq的重复性,比MNase-seq和DNase-seq的更强,操作起来也更加简便,而且只需要很少的细胞/组织量,同时测序信号更加好。目前已经成为研究染色质开放性首选的技术方法。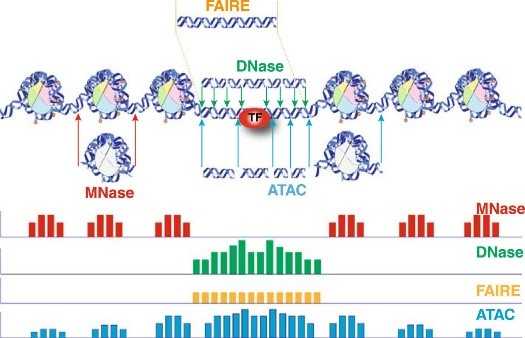
图2不同实验方法获得开放性染色质分析的示意图
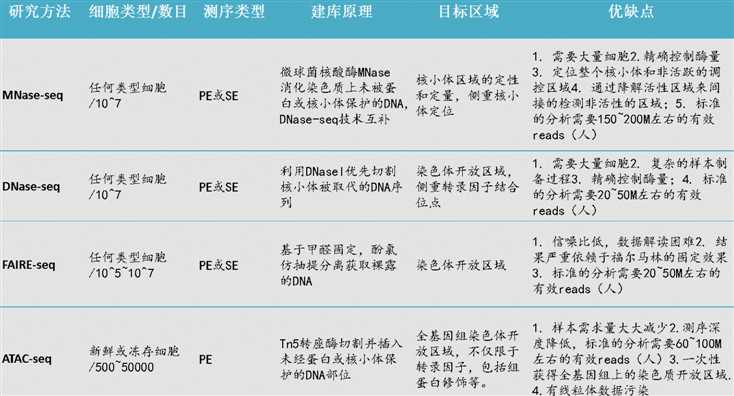
表1 ATAC-seq与传统方法比较
Q:ATAC-seq主要优势是什么?
A:面对现实需求ATAC-seq有以下四个方面的强大优势:
灵敏性高:低细胞起始量(500-50000个);
操作简单,耗时短;
实验重复性好:技术重复间表现出非常好的可重复性( R = 0.98) ,并与DHs 测序数据间也有着较好的一致性(R>0.79);
能同时揭示开放染色质的基因组位置,DNA结合蛋白,转录结合位点的相互作用
Q:ATAC-seq能用在那哪些方面?
A:结合ATAC-seq优势以及研究热潮ATAC-seq有以下用途;当然进一步的开发也可能在不久的将来让其使用更加广泛。
1. 非生物逆境,病虫害,营养,激素等处理前后及动物疾病,转录活性差异。
2. 不同组织,器官转录活性差异,找到组织特异基因和启动子。
3. 利用ATAC-seq技术来研究A、B、D三个亚基因组的转录因子结合位点的差异,从而研究同源基因的表达调控差异。而六倍体与二倍体、四倍体等位基因间调控位点的比较。
4 通过ATAC-seq定义的open chromatin区域 ,再结合motif 分析,识别哪种转录因子参与了基因表达调控(对于抗体质量不好的TF,尤其有效)
5 而将ATAC-seq和RNA-seq进行整合研究,将会获得对生物体(动物或植物)中的转录调控机制;宏观分析细胞在该特定时空下整个基因组的调控网络。
Q:ATAC-seq 有哪些缺陷呢?
A:任何技术都有其限制因素,ATAC-seq也不例外;
1. Tn5通过插入剪断DNA 并将测序接头连接到剪断的两个DNA 片段的末端,因此对于一个DNA 片段而言,其两端的接头连接是随机的,这便导致同一片段两端的接头有50%的概率是同一接头。而只有连接不同接头的片段才可用于富集扩增及测序,因此,有一半的片段无法利用;
2. 大量剪断的DNA 由于片段过大,无法进行PCR富集;
3. Tn5 的活性受反应溶液的组成及反应条件影响,仍然需要优化以便提高剪切效果;
4. ATAC-seq在植物细胞中存在以下难点:细胞壁的存在,叶绿体、线粒体等细胞器的污染,缺少稳定遗传的细胞系;
source: https://www.plob.org/article/13950.html
什么是染色质可及性/开放性?
真核生物中的核小体是染色质的基本结构单位。DNA与组蛋白结合后形成核小体,核小体再进一步折叠压缩后最终形成染色质。DNA的复制和转录都需要将染色质紧密结构打开,从而允许调控因子结合DNA。这部分被打开的染色质,就叫开放染色质(Accessible-Chromatin)。开放染色质允许调控因子结合的特性称为染色质的可及性(Chromatin Accessibility)。简而言之,染色质可及性/开放性是指某个时间点同时进行转录的DNA区域。
ATAC-seq实验原理
ATAC-seq利用DNA转座酶技术实现染色质可及性分析。DNA转座酶可以将自身结合的一段序列随机插入到基因组中。在ATAC-seq试验中,细胞或组织样本在核质分离后,将细胞核单独收集在一起,并通过转座酶对核内的染色质进行打断。紧密包裹的染色质DNA不会受到转座酶的打断,而开放区域的染色质DNA会被转座酶随机插入并打断。将这些打断后的DNA收集在一起进行后续的建库、测序、分析,即可得到开放染色质的信息。
ATAC-Seq可以解决什么问题?
转录因子的调控差异
ATAC-seq研究染色质的可及性,即某个时间点同时进行转录的DNA区域。这部分区域的染色质DNA被打开,因此可以被DNA聚合酶、转录因子、转录调控因子所识别、调控。在实验设计上,可以比较DNA聚合酶、转录因子、转录调控因子在不同类型样本中的转录调控差异。这些转录的差异又可以为下游的蛋白调控提供信息。
例如:在某些疾病、肿瘤组织,根据RNA-seq的结果提示样本转录表达的差异,在这些情况下,ATAC-seq可以从源头,即基因转录的情况提供信息,从而有可能证明转录差异的表达是由转录起始的某些调控因子引起的。
from
https://www.sohu.com/a/325903343_120055634
以上是关于如何使用ChromHMM鉴定染色质状态的主要内容,如果未能解决你的问题,请参考以下文章