Journal of Proteomics Research | 自动的可重复的免疫多肽数据分析流程MHCquant
Posted ilifeiscience
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了Journal of Proteomics Research | 自动的可重复的免疫多肽数据分析流程MHCquant相关的知识,希望对你有一定的参考价值。
题目:MHCquant: Automated and reproducible data analysis for immunopeptidomics
期刊:Journal of Proteome Research
发表时间:October 7, 2019
DOI:10.1021/acs.jproteome.9b00313
分享人:陈洁
本次分享的文章的题目是:MHCquant: Automated and reproducible data analysis for immunopeptidomics。
第一作者是图宾根大学的Leon Bichmann,通讯作者是Oliver Kohlbacher教授。
由HLA分子呈递在细胞表面的肽段在适应性免疫免疫中扮演着重要角色。T细胞识别HLA呈递的肽段并激活呈递肽段的细胞的死亡进程。HLA一类抗原决定簇是长度为8-12个氨基酸的多肽,是由一类抗原加工途径形成的内源性抗原。
来源于肿瘤特异性基因突变的新表位有希望作为靶标用于癌症免疫治疗。
为了从样本中更好的纯化和提取HLA配体,现已发展和优化了很多实验方法。相比之下,免疫信息学中几乎没有针对MS原始数据的处理方法,特别是针对HLA呈递的肽组(peptidome)。免疫多肽鉴定的一个大的缺点是它是非特异性酶切产生的,因此数据库搜索空间要远大于典型的胰蛋白酶酶切肽段的鉴定。又因为很大一部分免疫肽组来源于翻译后的剪切和非典型翻译,这就使得问题更为复杂。处理这样大的搜索空间和数据集会导致错误的鉴定,因此就需要稳健地估计和严格地控制假阳性率来防止错误的累积。此外,准确定量表位的丰度仍然是一个挑战。
随着鉴定这种肽段的需求越来越多,也发展出一些流程,但至今还没有集成的、版本控制的工作流程专门用于免疫多肽的鉴定。因此,作者开发了MHCquant,它是一个可用于鉴定和定量的流程。
- MHCquant流程的组成结构
MHCquant的处理步骤包括数据库搜索,FDR估计,非标定量和HLA结合亲和力预测。 FDR可以在多个水平(PSM,肽或蛋白质)上进行评估,可选地,这个软件可以通过计算高可信的和预测为亲和的PSM子集的FDR,从而挽救(rescue)低于常规FDR阈值的高可信PSM。此外,与其他可用的蛋白质组学软件相比,MHCquant将靶向特征提取作为一种非标定量方法,几乎可以对所有鉴定的肽进行完全定量,并可以在不同的run之间对比。
MHCquant是一个集成的数据处理管道(图1),包括来自OpenMS软件包的一系列不同的工具。这个流程的输入文件包括四类:一组LC-MS / MS原始文件(每个LC-MS / MS以mzML格式在单独的文件中运行),蛋白数据库(FASTA格式),突变调用文件(VCF格式),包含推定的新抗原突变和HLA分型文件(TSV格式)。使用Fred 2.0 免疫信息学框架将variants翻译成突变的蛋白序列,并添加到提供的FASTA数据库中。
该流程使用反序Decoy序列以Target-Decoy方法应用搜索引擎Comet进行数据库搜索。 随后,使用OpenMS的MapAlignerIdentification工具,将通过给定q值的共享的特征用于整个样本的线性保留时间对齐。 然后用Percolator重新计算FDR。
为了获得更好的FDR估计,MHCquant可以选择通过q值或通过给定的预测亲和力阈值的PSM作为一个子集重新计算其FDR。 最近,该技术已成功应用于宏蛋白质组学中的类似问题,并产生了优异的结果。
最后,结果以社区标准格式mzTab导出,并包括MHCflurry,MHCnugget与ImmunoNodes工具箱中其他的亲和力预测软件的预测结果。
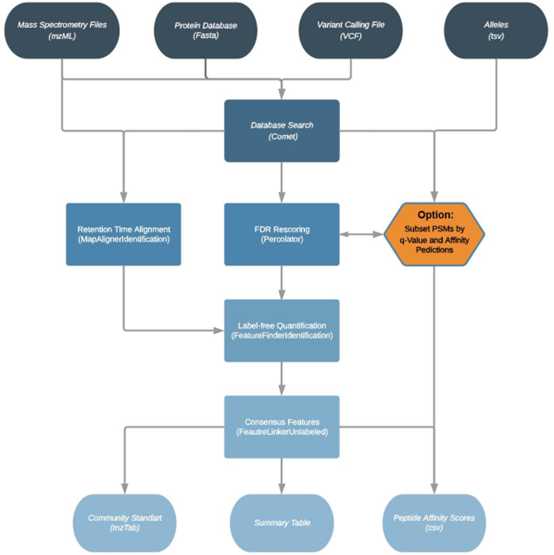
图1:多肽鉴定简化的工作流程
- MHCquant比现有方法具有更高的灵敏度
为了评估MHCquant的性能,作者使用一个包括9个PBMC样本和4个JY细胞系样本的数据集,比较了MS-GF +、PEAKS、SequestHT、Mascot和Andromeda的HLA -1结合多肽的鉴定结果。用q velue小于0.01且HLA亲和的肽段的数量和比率来评估每种搜索工具的性能。
在Subset FDR模式下,MHCquant鉴定到的HLA 亲和的unique peptide数最多,然后是默认模式下的MHCquant和PEAKS。所有搜索引擎鉴定到的肽都遵循预期分布,长度范围为8-12个氨基酸,最大长度为9。鉴定的HLA结合肽的结合率为87%至99%。
与其他工具相比,SequestHT,Mascot和MaxQuant鉴定到的亲和肽段的比例略高,而PEAKS的比率最低。相反,SequestHT,Mascot和MaxQuant鉴定到的HLA-1结合肽段较少。搜索引擎之间肽段的重叠表明,使用MHCquant和PEAKS可以鉴定到大多数unique peptide,并且这些unique peptide中预测亲和肽段的比率大于70%。此外,还比较了实际的保留时间与预测保留时间,结果显示预测的保留时间与所有共识鉴定的观察到的保留时间非常相关(r = 0.93)。总之,我们发现MHCquant在肽段鉴定数和HLA亲和肽段的比例之间提供了最佳平衡。
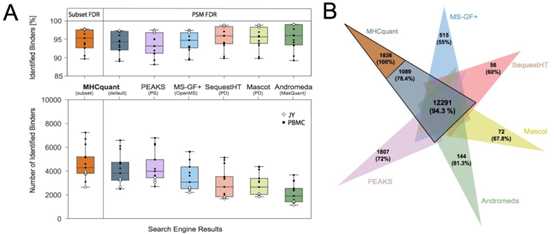
图2:各流程表现的比较
3.Subset方法在鉴定中的影响(增加12%的鉴定数)
通过subset 方法重新计算FDR后,鉴定到的unique peptide的中位数为4,000左右。与默认模式相比,以这种方式鉴定出的多肽中位数多了12%。
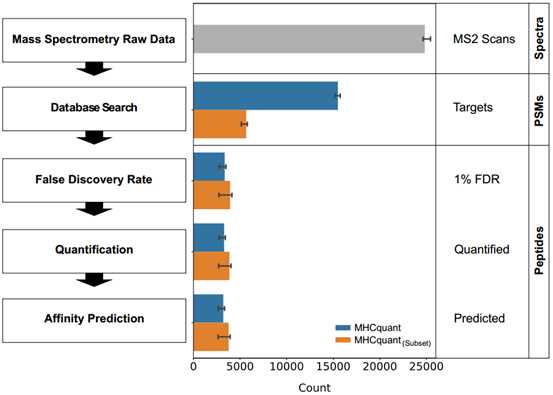
图3:MHCquant每个步骤中鉴定到的谱图或肽段的数量
(默认方式(蓝色)和subset FDR模式(橙色))
使用目标非标定量分析可以为99%的肽段定量。在从0.1 fmol到100 fmol的整个浓度范围内加到测量样品中的66种标记肽段中,有58种被成功鉴定和定量。 最终,取决于样品特性,预计约87%至99%的独特鉴定和定量肽是相应患者各等位基因的HLA-1结合肽段。
4.在旧数据中鉴定到新抗原
为了检查MHCquant相对于其他常用搜索工具的敏感性增加是否有助于在已发表的免疫肽组学数据集中发现新抗原,作者重新处理了近期的恶性黑色素瘤研究的质谱数据,鉴定结果中包含先前使用MaxQuant鉴定到的全部新表位。
表1:从已发表的黑色素瘤数据集中鉴定出新表位
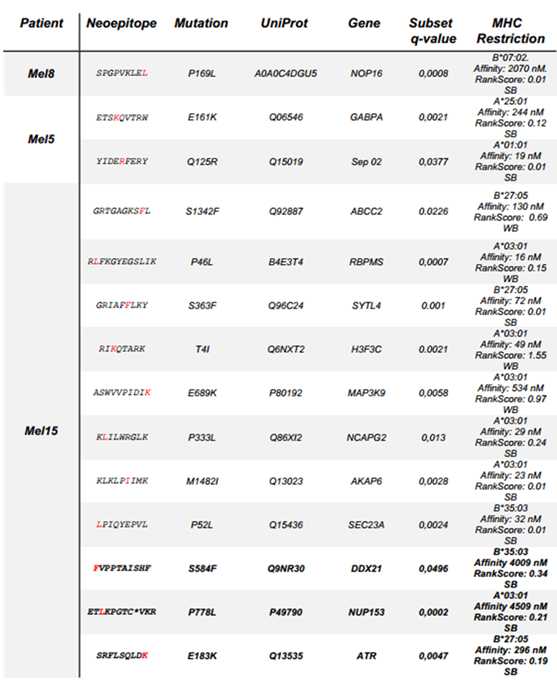
使用MHCquant,作者鉴定到了所有先前发表的新表位(表1)。此外还发现了三个潜在的新型潜在突变的新表位(表1加粗)。
通过与对应合成肽的谱图比较进行验证。所有三个肽段均显示出与先前检测到的新表位相似的范围。 其中一种肽片段(NUP153P778L)带有半胱氨酸氨基甲酰甲基修饰,这可能阻止了其在原始出版物中的检测。然而,氨基甲酰甲基修饰的肽仅占我们用MHCquant重新分析中所有鉴定出的肽的约2%。 总之,MHCquant允许以可重复的方式搜索免疫肽组学数据,其增加的敏感性可能会导致新发现。
小结:
MHCquant与其他的鉴定软件相比,可以更好地平衡肽段鉴定数和免疫亲和肽段的比例。一个关键的步骤是在搜索后把通过qvalue的肽段和通过预测的亲和力阈值的肽段作为一个新的集合重新计算FDR。
MHCquant在使用上有以下优点:一是该软件基于KNIME平台,是一个全自动、可移植的计算流程,在集群/云基础设施上执行和结果的完全重现性。
以上是关于Journal of Proteomics Research | 自动的可重复的免疫多肽数据分析流程MHCquant的主要内容,如果未能解决你的问题,请参考以下文章