RNA_seq 热图绘制
Posted zhanmaomao
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了RNA_seq 热图绘制相关的知识,希望对你有一定的参考价值。
若已经拿到表达矩阵exprSet
若差异较大,进行log缩小不同样本的差距
1、热图全体
1 ##加载包 2 library(pheatmap) 3 4 ##缩小表达量差距 5 exprSet <- log2(exprSet+1) 6 7 ##取最大标准差前1000个基因名字 8 cg <-names(tail(sort(apply(exprSet,1,sd)),1000)) 9 10 ##标准化,只关注样品间基因差异,不关注基因之间的,将离群点拉平 11 n <-t(scale(t(exprSet[cg,]))) 12 n[n>2] <-2 13 n[n<-2] <-2 14 15 ##这是group_list 16 group_list <-c(rep("a",3),rep("b",3)) 17 ac <- data.frame(sample=group_list) 18 rownames(ac) <- colnames(n) 19 20 ##draw pheatmap,其中annotation_col 可以增加sample组 21 pheatmap(n, show_rownames = F, show_colnames = F, 22 annotation_col = ac) 23 24 ##draw pheatmap add number 25 pheatmap(n, show_rownames = F, show_colnames = F, 26 annotation_col = ac, display_numbers = TRUE)
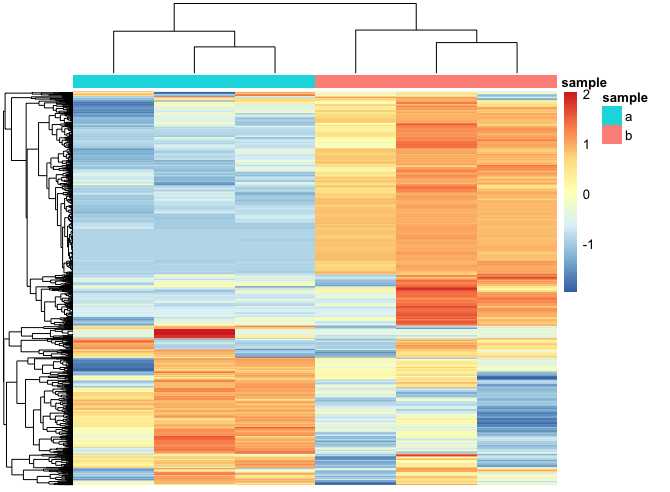
2、对差异基因进行绘制,步骤都类似,在进行绘制时,应对数据进行一定处理
1 ##DEseq2 获得dds 2 dds <- DESeq2(dds) 3 res <- results(dds) 4 res <- res[order(res$padj),] 5 DEG <- as.data.frame(res) 6 7 ##去掉NA 8 DEG <- na.omit(DEG) 9 10 ##热图 11 library(pheatmap) 12 choose_gene <- head(rownames(DEG),100) ##50 maybe better 13 choose_matrix <- exprSet[choose_gene,] 14 choose_matrix <- t(scale(t(choose_matrix))) 15 pheatmap(choose_matrix, show_rownames = F, show_colnames = F, 16 annotation_col = ac)
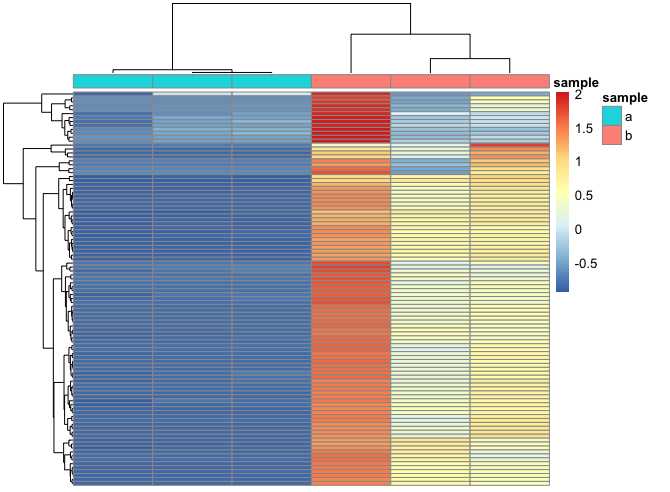
3、rlog 进行数据标准化
1 ##rlog 标准化 2 rld <- rlog(dds) 3 ##读取 4 exprMatrix_rlog <- assay(rld) 5 ##输出 6 write.csv(exprMatrix_rlog, ‘exprMatrix.rlog.csv‘)
标准化和raw count 的比较
png("DEseq_RAWvsNORM.png",height = 800,width=800) par(cex= 0.7) n.samples <- ncol(exprSet) if(n.samples>40) par(cex=0.5) cols <- rainbow(n.samples*1.2) par(mfrow =c(2,2)) boxplot(exprSet,col=cols, main="expression value",las=2) boxplot(exprMatrix_rlog,col=cols, main="expression value",las=2) hist(as.matrix(exprSet)) hist(exprMatrix_rlog) dev.off()
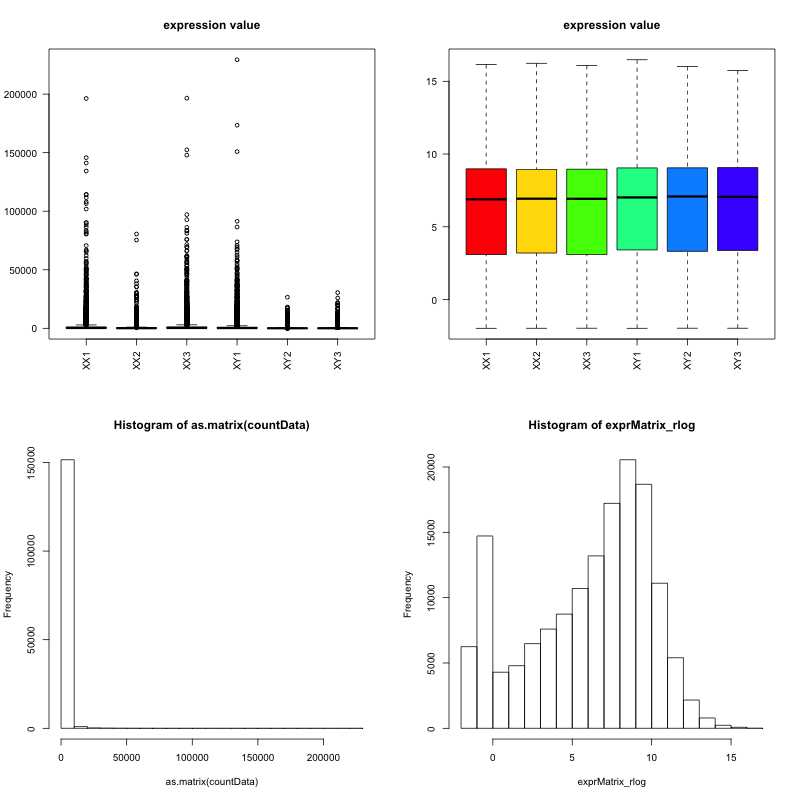
可以看到rlog后,样本间可以进行很好观察
参考:生信技能树
以上是关于RNA_seq 热图绘制的主要内容,如果未能解决你的问题,请参考以下文章