gff/gtf格式
Posted djx571
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了gff/gtf格式相关的知识,希望对你有一定的参考价值。
1.1)GFF3
GFF3允许使用#作为注释符号 ,除去注释外,主体部分共有9列。
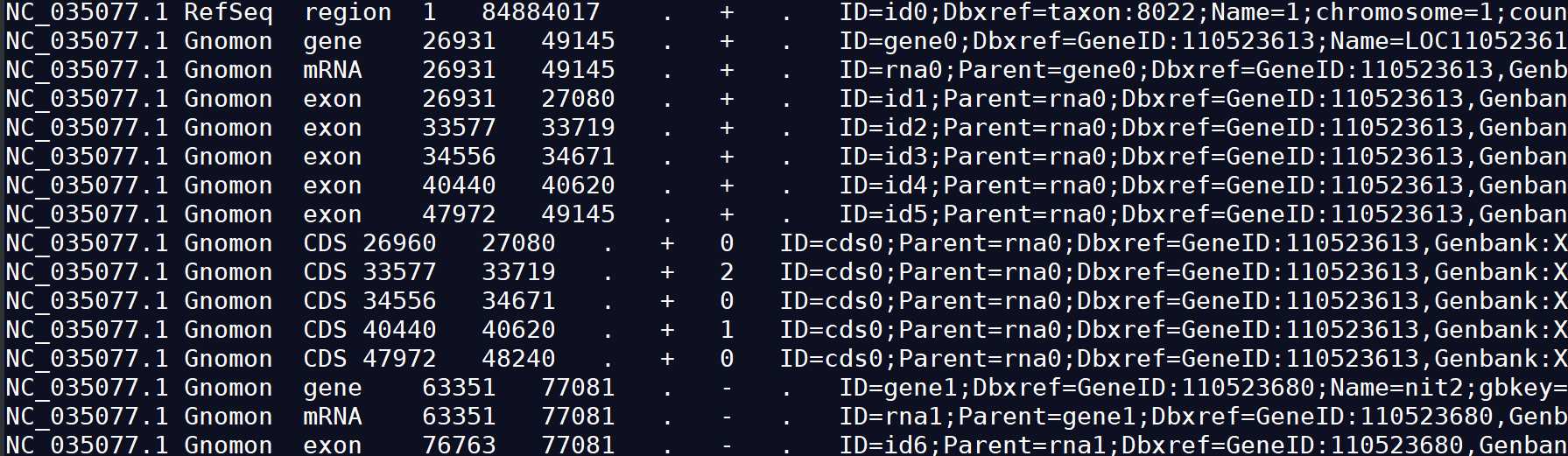
1) seqid :序列的id。(The name of the sequence where the feature is located.)
2)source:注释的来源,一般指明产生此gff3文件的软件或方法(e.g. Augustus or RepeatMasker)。如果未知,则用点(.)代替。
3)type: 类型,此处不受约束,但为了下游分析方便,建议使用gene,repeat_region,exon,CDS,或者是SO对应的编号等。
4)start:起始位置,从1开始计数(区别于bed文件从0开始计数)。
5)end:终止位置。
6)score:得分,是注释信息可能性的说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值。”.”表示为空。(indicates the confidence of the source on the annotated feature)
7)strand:“+”表示正链,“-”表示负链,“.”表示不需要指定正负链,“?” 表示未知.
8)phase :步进。仅对编码蛋白质的CDS有效,本列指定下一个密码子开始的位置。可以是0、1或2,表示到达下一个密码子需要跳过的碱基个数。
9)attributes:属性。一个包含众多属性的列表,格式为“标签=值”(tag=value),不同属性之间以分号相隔。
1.2)GTF2
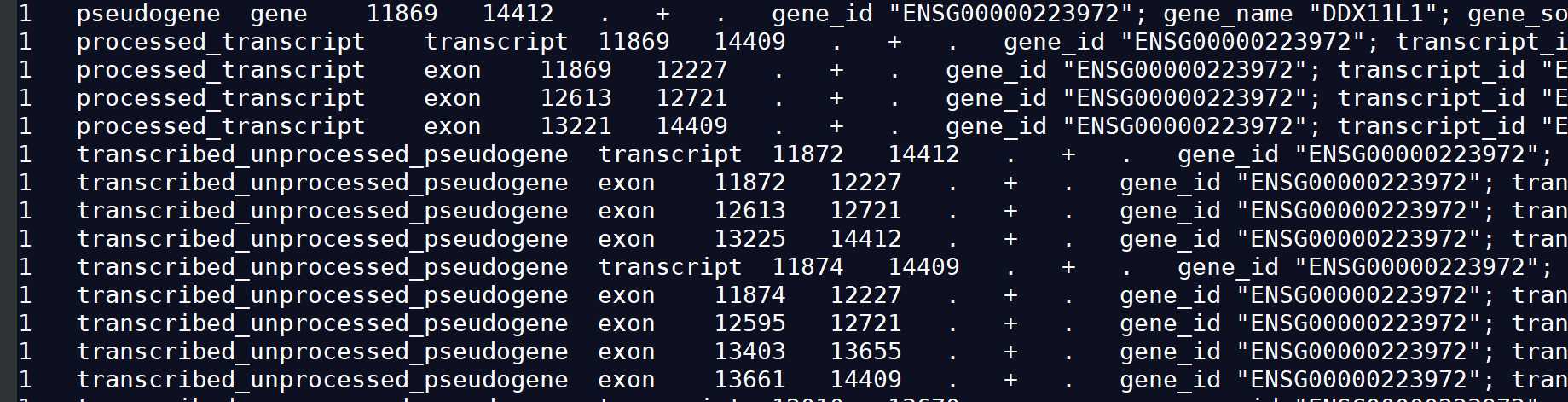
1) seqname: 序列的名字。通常格式染色体ID或是contig ID。
2) source:注释的来源。通常是预测软件名或是公共数据库。
3) start:起始位置,从1开始计数。
4) end:终止位置。
5) feature :基因结构.根据所使用的软件的不同,feature types必须注明。CDS,start_codon,stop_codon是一定要含有的类型。
6) score :这一列的值表示对该类型存在性和其坐标的可信度,不是必须的,可以用点“.”代替。
7) strand:链的正向与负向,分别用加号+和减号-表示。
8) frame:密码子偏移,可以是0、1或2。
9) attributes:必须要有以下两个值:
gene_id value: 表示转录本在基因组上的基因座的唯一的ID。gene_id与value值用空格分开,如果值为空,则表示没有对应的基因。
transcript_id value: 预测的转录本的唯一ID。transcript_id与value值用空格分开,空表示没有转录本。
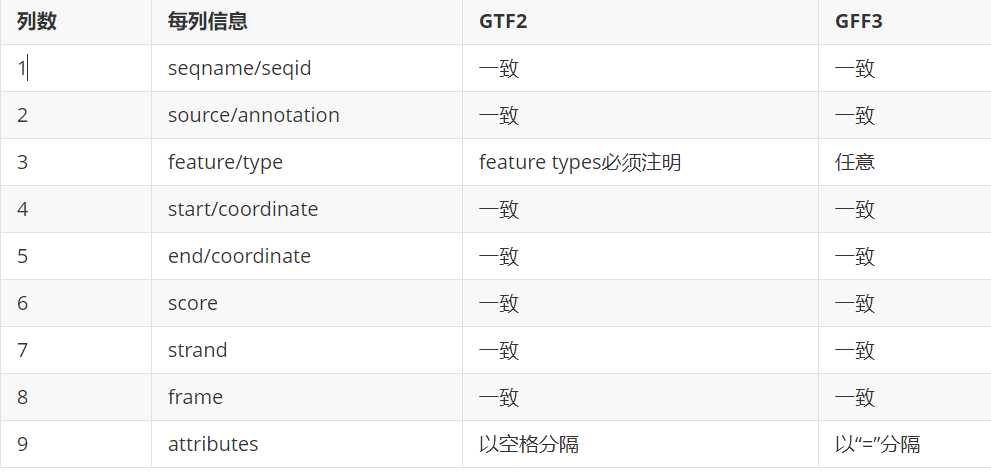
2)GFF3和GTF2之间的异同及相互转换。
gffread my.gff3 -T -o my.gtf #gff2gtf
gffread merged.gtf -o- > merged.gff3 #gtf2gff
以上是关于gff/gtf格式的主要内容,如果未能解决你的问题,请参考以下文章
如何在 Javadoc 中使用 @ 和 符号格式化代码片段?
SQLite 片段函数实现不会在 TextView 中将文本格式化为 HTML