生物信息学常识汇总
Posted univeryinli
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了生物信息学常识汇总相关的知识,希望对你有一定的参考价值。
电子病历方向
电子病历方面主要是筛选出冠心病影响的因素,通过抽取数据、数据缺失处理、数据文本校对、数据对齐等方式进行预处理,经过各种病的数据统计以后能够进行方向的确定,然后进行小样本分析,从而进行探索,相当于在机器学习这个层面还需要重新进行学习,感觉也会增加自己的时间成本。
- 心脑血管疾病致病因素
- 时序序列疾病的预测
多组学
基因组学,转录组学,蛋白质组学,代谢组学方面有很多的数据库可以做,而且基因组学和蛋白质组学方面的序列处理以及嵌入编码等方法与之前接触的词向量预训练模型有着相似的地方,从这个地方入手能够更快进入方向,将深度学习的技能和经验用到方法创新上面。
主要关注的方面为基因组学和蛋白组学方面
- 用神经网络对基因的表达量进行分类,有不同程度修饰的蛋白
- 染色质可及性和转录调控
- 从基因型数据预测基因表达的模型
- 鉴定lncRNA
- 研究单细胞中调控机制,如甲基化,亚型分析
- 基因组高级结构
- 基因组变异
- 基于长读长的数据利用深度学习进行base calling的技术
- 预测非编码元件变异的功能结果
- Nature Methods杂志上的一篇文章指出,DeepSEA 可以输入基因组序列,串联出大规模项目(如ENCODE和表观遗传学路线等)的染色质图谱,预测出一些重要调控位点的单核苷酸变异的影响,这些调控位点包括脱氧核糖核酸酶DNase敏感位点,转录因子结合位点,和组蛋白标记位点等
- DeepBind 能发现RNA与DNA上的蛋白结合位点,预测突变的影响。
- DeepVariant寻找基因变异,并且确定基因变异的位点,速度快,准确率高(谷歌)
首先要了解相关的基因方面的基本概念,包括基因结构、DNA结构、GWAS、SNP方面的结构等等。
基因的结构
DNA称为脱氧核糖核酸,可以组成遗传物质,一种由腺嘌呤脱氧核苷酸(dAMP )、胸腺嘧啶脱氧核苷酸(dTMP )、胞嘧啶脱氧核苷酸(dCMP )、鸟嘌呤脱氧核苷酸(dGMP )四种脱氧核糖核苷酸组成的长链聚合物
基因是DNA(脱氧核糖核酸)分子中含有特定遗传信息的一段核苷酸序列的总称,是具有遗传效应的DNA分子片段,是控制生物性状的基本遗传单位,是生命的密码,记录和传递着遗传信息。所有的基因都由4种碱基组成。
外显子和内含子,基因的编码区域里面包含外显子和内含子,外显子是直接可以转录成RNA的一段片段,内显子是经过修饰以后加入到转录的RNA中以后的片段,可以理解为内含子是外显子的补充。
基因的非编码区域,非编码区域占据基因片段的百分之90以上位点,在RNA的转录过程中并不发生转录行为,但是会控制编码区域的转录行为,比如启动子、终止子等等其他的附属功能都在这个区域,可以说这个区域是除了遗传信息意外的比较重要的区域,控制着编码区域基因的表达方式。
非编码区域与内含子的区别,既然内含子和非编码区域都不发生转录,那么肯定是有区别的,非编码区域只控制基因如何表达,比如基因的开始和结束,对于每一次转录他的作用都是一样的,并不会发生变化,存储着这一段基因特有的编码方式,但是内含子控制基因的编码内容,对于同一段基因不同时间的转录方式和RNA的组合方式,都会受到内含子的控制,可以说内含子虽然不直接进行编码,但是为基因片段在编码的时候提供了转录的多样性。
GWAS(Genome-wide association study),即全基因组关联分析,是指在人类全基因组范围内找出存在的序列变异,即单核苷酸多态性(SNP),从中筛选出与疾病相关的SNPs。通常与疾病相关的SNP变异大多不是在编码蛋白质的DNA区域,相反,他们通常位于非编码区域上,或者位于编码基因的内含子上面,虽然这个变异不直接进行基因的编码,但是是可以控制外显子表达的重要基因片段。由于GWAS研究的各种研究设计方法以及遗传统计方法无法从根本上消除人群混杂、多重比较造成的假阳性,我们需要通过重复研究来保证遗传标记与疾病间的真关联。
简单来说,就是将基因测试人员分成两组,一组为case组,一组为control组,分别对相同位置的snp位点计算同组内所有人的的cIBD得分,每个人都相对于其他人计算得分值,然后比较两组得分的差异,差异比较大的snp为变异点,这不利于筛选多个位点的变异,变异其实就是当前个体相对于其他所有个体的差异性,现在的工作基本都是通过基因层面来数值化分析snp位点的差异,并不是通过变异位点的编码序列来判定位点的变异,通过基因序列的差异性变化能够分析出多个基因的差异性,能够更加准确得判定序列的差异了,而且容易生成自动化方案。
比如,寻找糖尿病的致病基因是哪一个位点,可以找到乳腺癌的致病SNP是那些,等等
mRNA,为messenger RNA 的简称,或称为信使RNA。mRNA是由DNA经由转录而来,带着相应的遗传讯息,为下一步转译成蛋白质提供所需的讯息。在细胞中,mRNA从合成到被降解,经过了数个步骤。在转录的过程中,第二型RNA聚合酶(RNA polymerase II)从DNA中复制出一段遗传讯息到mRNA前体pre-mRNA(尚未经过修饰或是部份经过修饰的mRNA,称作pre-messenger RNA,pre-mRNA,或是heterogeneous nuclear RNA,hnRNA)上。
MicroRNAs(miRNAs)是一种小的内源性非编码RNA分子,大约由21-25个核苷酸组成。这些小的miRNA通常靶向一个或者多个mRNA,通过翻译水平的抑制或断裂靶标mRNAs而调节基因的表达,通过与mRNA结合控制基因表达的程度和水平。
miRNA-mRNA的结合预测,不同的miRNA控制着不同给表达程度,通过分析两个序列的序列信息可以对基因表达的抑制程度进行预测,还可以分析出来是哪些位点的结合使他有着不同的表达程度,下面有着预测的数据库.
等位基因和非等位基因,在一对同源染色体的同一位置上控制着相对性状的基因,非等位基因是位于非同源染色体上或同源染色体的不同位置上控制着不同性状的基因。等位基因之间存在相互作用。当一个等位基因决定生物性状的作用强于另一等位基因并使生物只表现出其自身的性状时,就出现了显隐性关系。作用强的是显性,作用被掩盖而不能表现的为隐性。一对呈显隐性关系的等位基因,显性完全掩盖隐性的是完全显性(complete dominance),两者相互作用而出现了介于两者之间的中间性状。等位基因的相互作用和非等位基因的相互作用。等位基因的相互作用表现为显隐性关系,而非等位基因的相互作用表现统称为上位效应。等位基因的差别可能是因为一个或者多个SNP导致等位基因差异,也有可能只是因为一个SNP差异导致。
SNP和SNV的区别,SNP(Single Nucleotide Polymorphisms)是单核苷酸多态的简称,SNV(Single Nucleotide Variant)是指单核苷酸结构变异,如果在一个物种中该单碱基变异的频率达到一定水平就叫SNP,而频率未知(比如仅仅在极少数个体中发现)就叫SNV。
基因填充
基因型填充在现在的全基因组分析中扮演着重要的作用,因为在测量基因的时候,因为基因芯片的原因,会丢失一些基因,所以不同个体的基因测序的数量是不一样的,这对我们的基因分析带来一定程度上的困难,所以基因型填充很必要。
基因型填充可分为两大类,一类是家系数据中的基因型填补,另一类是无关个体中的基因型填补。家系数据中的基因型共享染色体比较长,包含数千个SNP,而无关个体中共享染色体区域比较短,使得寻找匹配的单倍型成为一个挑战。
基因型填充方法,期望最大化算法(EM),马尔可夫链-蒙特卡洛算法,聚类算法,因马尔可夫算法。
基因型填充的软件:准确度优先,就是在填补基因的时候考虑每个缺失基因和所有位点的关系,这种方法所耗费的时间比较长,但是准确率高;另外一类方法是根据缺失位点附近的已分型位点来进行填补,这种方法计算量会减少,但是也牺牲了一部分正确率。
Assessment of factors affecting imputation accuracy,影响基因填充精度的因素,The SNP Density, sample size, and minor allele frequency of the SNP
Linkage Disequilibrium,计算差异不均衡是评价变异基因位点之间关系的一个评价,这个概念比较老,其实就是
生物信息中英文对照
Indel-插入缺失,chromosome-染色体,exome-外显子组,whole genome sequence-WGS全基因组序列,intron-内含子,biallelic-等位基因,recalibration-再校准,low coverage - Low coverage whole genome sequencing-低通量测序全基因组序列,exome - Whole exome sequencing-全外显子序列,high coverage - PCR-free high coverage whole genome sequencing-高通量全基因组序列,variation-变异,contig-重叠序列,Panel-面板,alleles-等位基因,trio-sWGRs-家系全基因双,trio-sWGs-家系准全基因组,Linkage Disequilibrium-差异不均衡
基因数据库-1000 Genome
基因组官方网站
NCBI官方网站基因浏览器
主要是SRA数据集,主要的优点是可以浏览,并且能够根据浏览的基因通过[SRA toolkit下载] (https://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software)
基因组数据库:ftp://ftp-trace.ncbi.nih.gov/1000genomes/
推荐比较好的基因组博客:https://www.plob.org/tag/sra/
http://www.bio-info-trainee.com/
1000 Genomes Project(缩写为1KGP)于2008年1月启动,是一项国际研究工作,旨在建立迄今为止最详细的人类遗传变异目录。科学家计划在接下来的三年内使用新开发的技术对来自不同种族群体的至少一千名匿名参与者的基因组进行测序,这些技术更快,更便宜。 2010年,该项目完成了试验阶段,在“自然”杂志的一篇出版物中对此进行了详细描述。2012年,1092个基因组的测序在Nature出版物中公布。 2015年,“自然”杂志上的两篇论文报告了结果,项目的完成以及未来研究的机会。确定了许多罕见的变异,仅限于密切相关的群体,并分析了8个结构变异类别。
这里面有多个数据模式,有原始数据和分析处理以后的数据
- 原始数据-fastq,原始数据是直接从基因芯片得到的数据,是没有经过Align的基因序列,文件格式为fastq格式,Linux可以通过zcat打开,也可以通过cat打开,每一行数据的分隔符为 进行分割,原始数据的fastq文件大小大概为2g大小,所在的位置为ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/1000genomes.sequence.index,具体的图片为下图
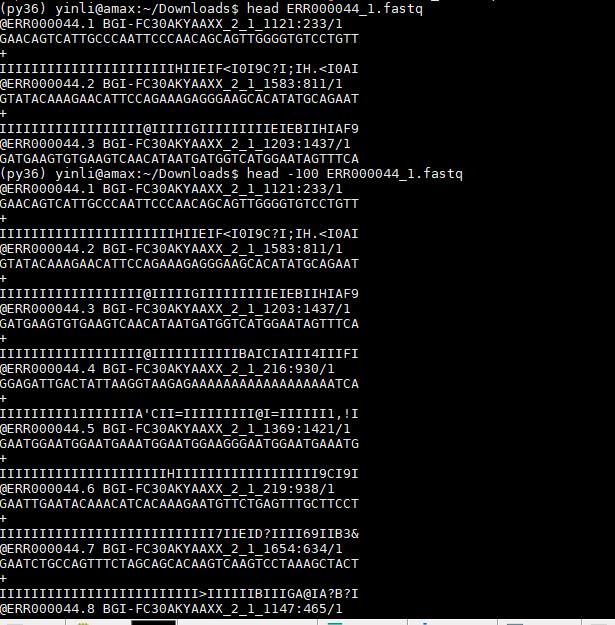
fastq格式是生物信息分析中最常见的格式之一,通常测序的数据分为双端测序和单端测序,双端测序的数据含有两个fastq格式的文件,单端测序的数据只有一个fastq格式的文件,1000 g数据都包含两个fastq文件属于双端测序
fastq文件格式主要分四行:
第一行是用来区分不同reads的一个ID号,一般以@符号开头,这一行是用来区分不同的reads,而这一行本身包含了很多的信息。Read Record Header,Flow Cell ID,Lane,Tile,Tile Coordinates,Barcode
第二行是测序的序列,也就是reads的序列
第三行一般是一个+号,或者与第一行的信息相同
第四行是碱基质量值,是对第二行序列的碱基的准确性的描述,一个碱基会对应一个碱基质量值,所以这一行和第二行长度是一样的,如果不一样就说明数据有问题,这一行的质量值是通过ACII码来说明的,将码进行转换就可以得到分数值,ACII码转换为质量百分值的过程为,Q=-10 log10p标准,或者 Q=-10 log10p/(1-p)标准,两种计算方式在高质量的时候没有差别,在低质量的时候差异明显
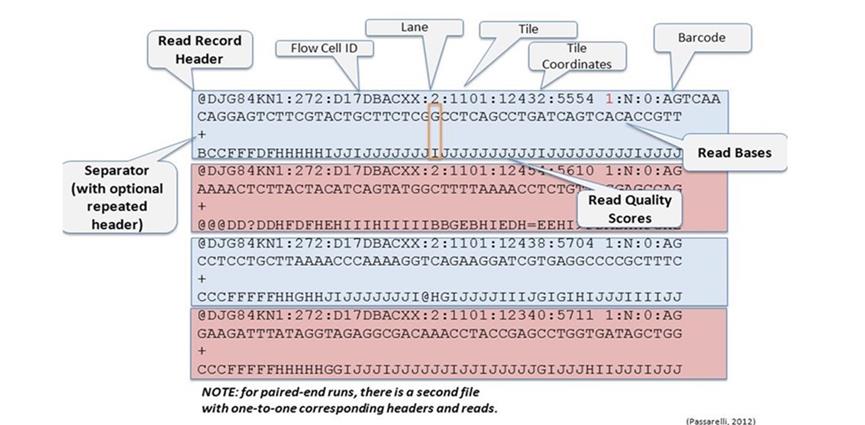
Fastq格式的解析细节可以参考该博客:https://www.cnblogs.com/djx571/p/9493934.html
- align的基因序列-cram格式文件
包含三个文件夹,分别为低通量全基因序列、高通量全基因序列、全外显子序列,都是fastq文件经过比对和对齐来产生的,
- cram是sam文件的压缩版本,有着很多优点,在保证信息完整的情况下可以将压缩率加大,使文件变得更小,cram文件结构
- bam则是sam的二进制版,在sam的基础上运用二进制编码,又极大的压缩了sam文件的体积。
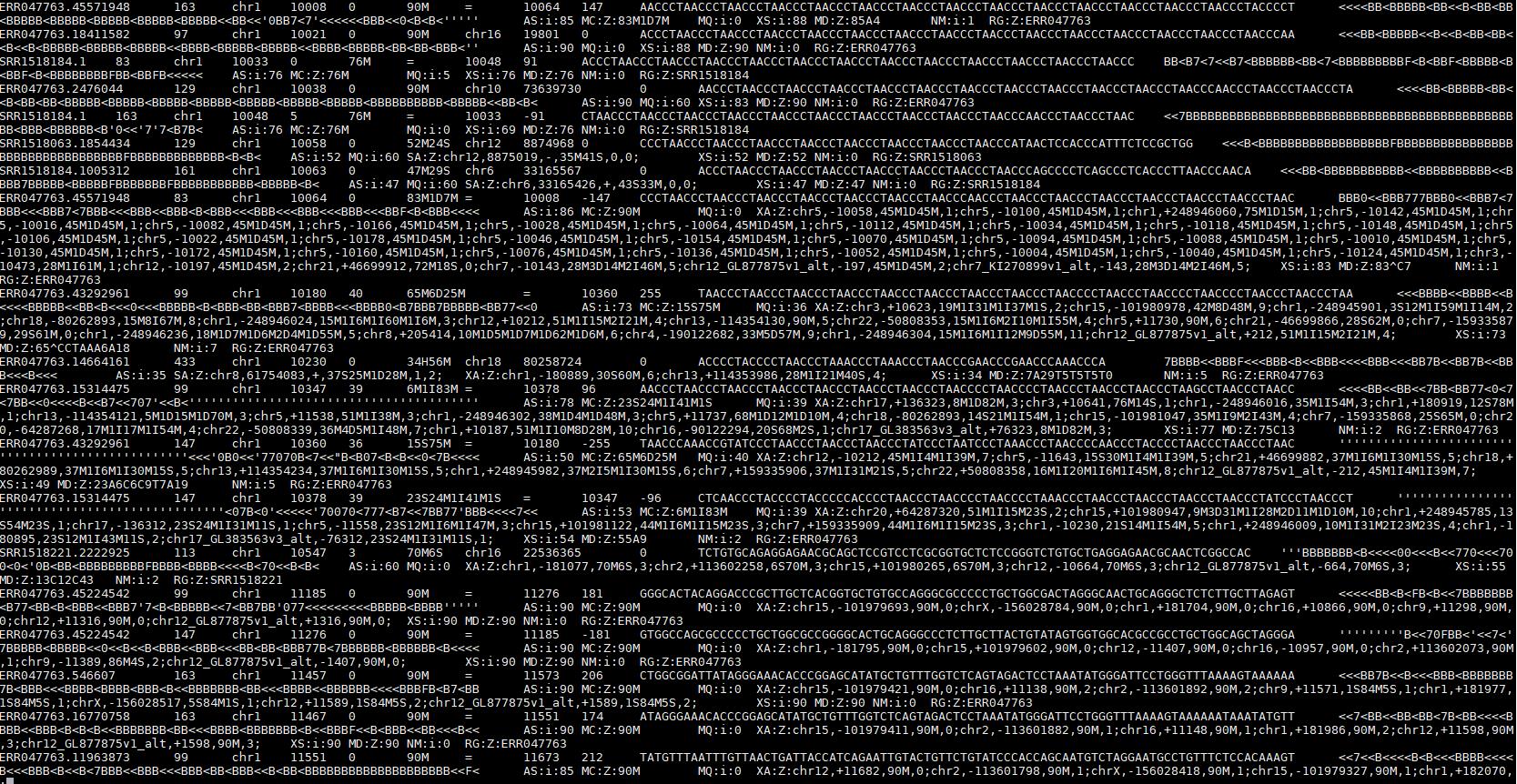
SAM文件主要由两个部分构成
header:标记了该SAM文件的一些基本信息,比如版本、按照什么方式排序的、Reference信息等等。
本体:每行为一个reads,不同列记录了不同的信息,列与列之间通过tab分隔。

QNAME:测序的reads的名字。
FLAG:二进制数字之和,不同数字代表了不同的意义;比如正负链,R1/R2(双端测序的哪一端)等。
RNAME:map到参考基因组后的染色体名称。
POS:1-based 基因组起始位点。
MAPQ:map的质量。
CIGAR:一个数字与字母交替构成的字符串,标记了这段reads不同位置的match情况。不同字母的含义后边介绍。
RNEXT:如果是pair-end测序,这个为mate(另一端中对应的)的read的染色体名称;否则为下一条read的染色体名称。
PNEXT:同上,read对应的起始位点。
TLEN:模板的长度。
SEQ:序列。
QUAL:序列的质量打分(fasta文件中的那个)。
更加详细的文件结构说明请参考博客总结,该博客总结的比较好:https://www.jianshu.com/p/a584d31418f3
- 基因分析文件-vcf文件
VCF是用于描述SNP(单个碱基上的变异),INDEL(插入缺失标记)和SV(结构变异位点)结果的文本文件。在GATK软件中得到最好的支持,当然SAMtools得到的结果也是VCF格式,和GATK的CVF格式有点差别,GATK是一款分析SNp变异位点的软件。
生物基因数据文件-博客非常详细得解释了vcf文件的组成结构:https://blog.csdn.net/u012150360/article/details/70666213
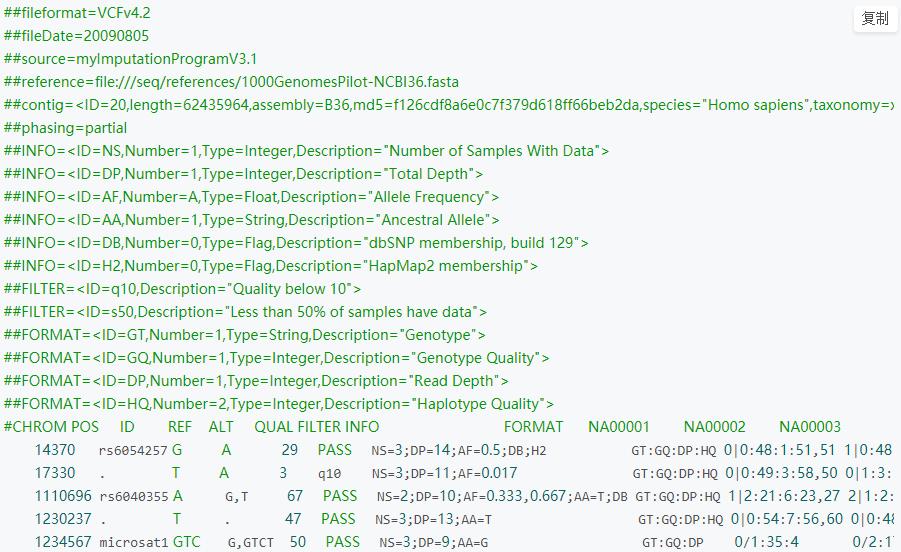
GATK 是 Genome Analysis ToolKit 的缩写,是一款从高通量测序数据中分析变异信息的软件,是目前最主流的snp calling 软件之一。GATK 设计之初是用于分析人类的全外显子和全基因组数据,随着不断发展,现在也可以用于其他的物种,还支持CNV和SV变异信息的检测。在官网上,提供了完整的分析流程,叫做GATK Best Practices。主要识别SNP和CNV 两大类型的变异,每种变异类型又有Germline和Somatic的区别。通过GATK分析以后的文件类型为vcf为表格数据,通过excel或者pandas可以直接读取,vcf中存储数据为所有的变异位置和位点信息。
Germline指的是在胚胎发育早起出现的变异,这种变异会在所有细胞中广泛存在,是可以遗传给后代的变异;Somatic指的是体细胞变异,身体特定区域或者组织中出现的变异。通常不会遗传给后代。
全外及全基因双组学遗传突变分析
从检测范围上看:
Panel,部分基因的组合,一般是由几百个基因组成的DNA序列,这样的分法是在全基因组比较昂贵的时候进行分的,现在价格比较低了,往往直接测整个基因组的序列的就可以,不用单独针对一部分疾病的区段进行panel测序
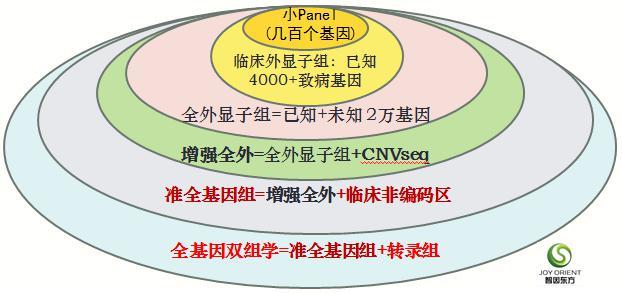
家系准全基因组(trio-sWGsTM),是在增强全外的基础上,增加了对人类全部四千多种致病基因的非编码区的trio(一家三口)测序,可以检测到近全部非编码区的已知致病突变。虽然这不是标准意义上的全基因组30X测序(WGS),但在致病突变相对密集的全外显子和临床非编码区的区域可以获得100X测序深度,其数据质量要远胜于WGS。
家系全基因双组学(trio-sWGRsTM),是在家系准全基因组的基础上,增加了一家三口的外周血白细胞全转录组测序(RNAseq),可以检测分析白细胞表达的近万个基因的表达谱和各种剪接变体。对于致病基因在白细胞表达并发挥功能的一些疾病,尤其是血液系统疾病、免疫系统疾病及一些大分子代谢疾病等,全基因双组学策略不仅可以检出已知的非编码区致病突变,还有可能检出新的致病变异,而且能够得到变异在转录组层面的功能验证(比如影响调控表达、影响剪接等)。
从检测模式上看:
这里说的检测模式是指先证者模式(即二代测序只测先证者,挑出怀疑变异再做一代验证),还是核心家系(trio)模式(即二代测序同时检测先证者和父母)。对于全外显子、全基因组如此大的检测范围,只检测先证者是不可取的。因为先证者模式无法判断变异是否呈现家系共分离,即便挑几个怀疑的变异去做家系一代验证,也很容易挑错,漏掉真正的致病突变。
先证模式还有一个坑,那就是即便挑了少数变异去做家系验证,但也无法知道父母样本是否来自真正的生物学父母,而trio模式则可以借助大数据比对来判断生物学父母的可靠性。
trio-WES,或称核心家系全外显子组测序,已成为目前遗传病诊断的基本配置。在此基础上再增加核心家系的CNVseq、临床非编码区、转录组,就分别成为更为强大的增强全外家系、家系准全基因组、家系全基因双组学策略。
从适用变异形式上看:
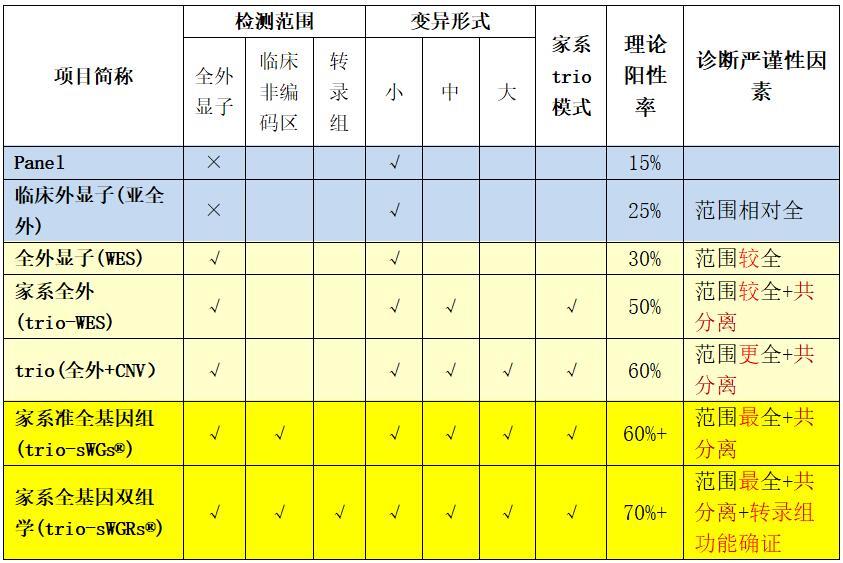
遗传病的基因序列变异主要可以分小(点突变)、中(基因及内部外显子的缺失重复)、大(100kb以上大片段CNV)这三类。
一般的Panel,临床外显子组,甚至全外显子组,只能检测小型变异,对中型和大型变异无法检出,也就是若在Panel范围内的某个基因及其相关区域存在致病的CNV,会大概率漏检。
智因的trio-WES,可以利用家系全外显子数据,对全部约2万个基因进行外显子缺失重复的筛查,同时实现小型和中型变异的检出。传统的中型变异检测方法是MLPA,其局限性是只能检测指定某一个基因是否存在外显子缺失重复,而智因全外可以全面扫描几乎全部2万个基因的外显子缺失重复。如果把MLPA比喻为“狙击点射”,则智因全外的中型变异筛查则是“地毯式轰炸”。全外分析对连续两个以上的外显子缺失重复的准确性较高,但对单个外显子拷贝数异常的检测准确度不及MLPA,如果医生强烈怀疑某个基因的问题,可以单加这个基因的MLPA检测。二者各有优缺点,不必相互菲薄。
智因的trio(WES+CNVseq),或trio(全外+CNV),或称家系增强全外,在WES的基础上,增加了全基因组CNVseq检测,不仅可以弥补大片段拷贝数变异的检测,而且还能得到单亲二倍体(UPD)的检出,即可以全面涵盖大中小三类变异。CNVseq方法已得到大样本的验证,其灵敏度和特异性与CMA芯片一致率。
综合对比几种检测策略:
数据格式解读
有一些公共的基因填充网站,已经做好开源网站:
https://imputationserver.sph.umich.edu/index.html#!pages/home
网站的使用说明:
https://www.cnblogs.com/chenwenyan/p/10830207.html
samtools软件安装
官方软件地址:http://www.htslib.org/
GitHub:https://github.com/samtools
Samtools软件是一个能够读取SAM/BAM/CRAM的套件,同时也能够读取fastq等一系列基因文件,BCFtools是能够处理BCF2/VCF/gVCF等文件的套件,两个都依赖HTSlib库。
Linux一般软件都可以使用sudo apt install进行安装,但是该软件需要使用本地编译安装使用,下载samtools软件,然后安装一下步骤安装
检查安装所需要的包是否完整,如果不完整需要先安装其他的包
./configure
#编译可执行文件
Make
#对可执行文件进行安装
make install
samtool所依赖的部分包地址,按照地址下载,并且安装上面的方法进行安装就可以,如果缺少什么软件,那么就再去安装所需要的依赖,不过安装的都是lib文件,所以不会出现二次依赖的问题
Samtools and HTSlib depend on the following libraries:
Samtools:
zlib http://zlib.net
curses or GNU ncurses (optional, for the ‘tview‘ command)
http://www.gnu.org/software/ncurses/
HTSlib:
zlib http://zlib.net
libbz2 http://bzip.org/
liblzma http://tukaani.org/xz/
libcurl https://curl.haxx.se/
(optional but strongly recommended, for network access)
libcrypto https://www.openssl.org/
(optional, for Amazon S3 support; not needed on MacOS)
Linux上采用samtools进行软件分析比较方便,但是用python又是一个问题了,通过安装pysam可以解决这个问题,Linux下通过指令就可以直接安装
pip install pysam
pysam综合了htslib的所有功能,能够对SAM/BAM/VCF/BCF/BED/GFF/GTF/FASTA/FASTQ等格式的文档进行操作处理,为python处理基因数据提供了很好的辅助工具
pysam使用文档:https://pysam.readthedocs.io/en/latest/index.html#
VCFtools的使用
VCFtools主要是用来打开vcf等文件的,同时进行snps的分析等操作,可以通过Linux等进行下载,其下载和说明文档的地址:
https://vcftools.github.io/index.html
生物医学文本挖掘
-
基于深度学习的生物医学命名实体识别,深度学习的方法对医学命名实体进行提取,包括生物信息学(有一定的公开数据集,主要是基因,蛋白,DNA,RNA等方面文本数据)
-
生物医学文本挖掘-利用文本特征用于提取文献中药物之间的关系(有一定数量公开数据集)
https://blog.csdn.net/SA14023053/article/details/45667031 -
生物医学文本挖掘BioNLP-自动提取出复杂的生化反应网络
https://blog.csdn.net/yanqianglifei/article/details/80486623
以上是关于生物信息学常识汇总的主要内容,如果未能解决你的问题,请参考以下文章